Fabrijeva bolest
Fabrijeva bolest (akronim FB) ili Anderson–Fabrijeva bolest predstavlja za X vezano, genetski uslovljeno oboljenje lizozima kod koje dolazi do nakupljanja glikosfingolipida na unutrašnjem sloju krvnih sudova (endotelu), na brojnim unutrašnjim organima, na njihovim epitelima i glatko-mišićnim strukturama.[1]
| Fabrijeva bolest | |
|---|---|
| Sinonimi | Fabry's disease, Anderson–Fabry disease, angiokeratoma corporis diffusum, alpha-galactosidase A deficiency |
 | |
| Alfa galaktozid - deficitaran proteina kod Fabrijeve bolesti | |
| Izgovor | |
| Specijalnosti | endokrinologija, kardiologija, nefrologija, dermatologija |
| Komplikacije | oštećenje srca, srčane aritmije |
| Vreme pojave | detinjstvo |
| Uzroci | genetika |
| Dijagnostički metod | test aktivnosti enzima, genetsko testiranje |
| Lečenje | zamena enzima |
Fabrijeva bolest je tipičan predstavnik retkih bolesti, koja za razliku od mnogih drugih trenutno ima mnogo povoljniju prognozu, nakon uvođenja supstitucione enzimske terapije i razvoja boljih kliničkih, biohemijskih i molekularnih dijagnostičkim metoda koje mogu značajno da skrate vreme prepoznavanje ovih pacijenata.[2]
U zavisnosti od organa koji su zahvaćeni, mogu se pojaviti veoma različiti simptomi. Pojedinačno veoma različita manifestacija bolesti i njena retkost znatno otežavaju dijagnozu , obično se ispravno postavlja tek mnogo godina nakon pojave prvih simptoma.
Bolest prvenstveno pogađa muškarce, ali heterozigotne (mešane krvi) žene takođe mogu da obole. Kod njih je, međutim, bolest obično manje teška i tek u srednjim godinama počinje da postaje klinički relevantna. Kvalitet života pacijenata koji pate od Fabrijeve bolesti je često dosta oštećen.
Bolest se uzročno leči terapijom zamene enzima od 2001. Pacijenti dobijaju genetski modifikovanu α-galaktozidazu A kao infuziju do kraja života . Trenutno ne postoji lek za Fabrijevu bolest. Ako se ne leče, muški pacijenti žive do prosečne starosti od oko 50, a žene do oko 70 godina. Glavni uzroci rane smrtnosti su hronična bubrežna insuficijencija , oštećenje srca i oštećeno dotok krvi u mozak .
Aktivnost enzima α-galaktozidaze A je u velikoj meri smanjena mutacijom ( genetskom modifikacijom) na H hromozomu (polni hromozom) , odgovoran je za razgradnju masti. U lizozomima (centri za reciklažu ćelija), metabolički proizvod globotriaozilceramid (koji se takođe naziva Gb3 ili GL-3), glikosfingolipid , više ne može da se razgradi u dovoljnoj meri. Gb3 se akumulira prvenstveno u ćelijama koje oblažu unutrašnjost krvnih sudova , endotelnim ćelijama, pa kako bolest napreduje, ove akumulacije postaju patološke , odnosno izazivaju Fabrijevu bolest. U zavisnosti od toka bolesti, to ponekad može potrajati decenijama.
Istorija uredi
Kao poseban klinički entitet, Fabrijevu bolest je prvi put opisao engleski hirurg Vilijam Anderson 1897. godine.[1]
Godinu dana kasnije (1898. godine), nemački dermatolog Johan Fabri je opisao difuzne angiokeratome u 13 godina starog dečaka sa proteinurijom, koji su takođe bolovali od ove bolest, pa je je ovaj eponim po imenu Johanesa Fabrija nazvan Fabrijeva bolest,[1] mada se u literaturi ova bolst naziva i Anderson-Fabrijeva bolest.[3][4]
Epidemiologija uredi
Učestalos Fabrijeva bolesti se kreće od 1:50 000 (1:40 000 do 1:117 000) iako su blaži oblici bolesti koji se javljaju kasnije u životu javljaju nešto češće.[5][6][7] Međutim, novije studije zasnovane na podacima sa skrininga novorođenčadi ukazuju na značajno veću učestalost Fabrijeve bolesti. Od 2004. do 2006. godine, prevalencija od oko 1:3.100 na Tajvanu[8], a oko 1:1.500 kod muške novorođenčadi u severnoj Italiji, ili sa prevalencom od 1:3.100, koja rezultuje sa 26.450 pacijenata sa Fabrijevom bolešću u Nemačkoj.[9]
Pretpostavlja se da kod mnogih pacijenata bolest ostaje neprepoznata tokom njihovog života i da se prevremena smrt pripisuje drugim bolestima, kao što su izolovane kardiomiopatije poput onih koje se nalaze kod pacijenata sa Fabrijevom bolešću sa rezidualnom aktivnošću α-galaktozidaze A.[8] Još jedan pokazatelj ovoga je da je na Tajvanu 86% novorođenčadi koja su bila pozitivna imala kriptičnu mutaciju spajanjatipa IVS4+919G > A, koji je ranije pronađen prvenstveno kod pacijenata sa Fabrijevom bolešću sa srčanim fenotipom.[10] Kod ovih pacijenata, Fabrijeva bolest se u suštini manifestuje bolešću miokarda (kardiomiopatija). Intronički oblik ove mutacije prisutan je kod mnogih tajvanskih pacijenata sa hipertrofičnom kardiomiopatijom.[11][12]
U određenim subpopulacijama povezanim sa simptomima Fabrijeve bolesti, incidencija je sama po sebi veća. Studija na 911 španskih pacijenata na hemodijalizi identifikovala je četiri muškarca i tri žene sa abnormalnostima u GLA genu, što odgovara prevalenciji od 1:182 u ovoj subpopulaciji.[13]
Etiologija uredi
Fabrijeva bolest je za H vezana nasledna lizozomska bolest deponovanja zasnovana na lizozomskom nedostatku enzim alfa galaktozidaza (AGAL-A) odgovornog za hidrolizu galaktoze ostatka sfingolipida zbog čega su oni (poput globotriazilceramida, galabiozil ceramida i globotriazilsfingozin, sfingozin 1 fosfat) akumuliraju u brojnim ćelije i tkiva.[1] Ova akumulacija ostatka sfingolipida rezultuje progresivnim oštećenjem tkiva i organskim oštećenja koja su najčešće posledica promena u:[3][1]
- endotelu i glatkih mišićnih ćelija mikrocirkulacije,
- akumulaciji metabolita sfingolpida u ćelijama srca, bubrega, rožnjače, pankreasa, creva, pluća, pankreasa i
- raznih vrsta nervnih ćelija centralnog i perifernog nervnog sistema.[14]
Genetika i molekularna biologija uredi


A Sedamnaestogodišnja devojka sa transverzijom timina u gvanin u eksonu 6, pozicija 884. Ova supstitucija nukleotida menja kodon TTC, koji kodira aminokiselinu fenilalanin , u TGC, što se prevodi u inkorporaciju cisteina u proizvod gena α-galaktozidaze.
A. Shodno tome, u α-galaktozidazi A ovog pacijenta, nalazi se cistein na poziciji 295 umesto fenilalanina. Postoji mutacija p.Phe295Cis.
B. 46-godišnja žena sa T-to-G transverzijom u svom GLA genu na egzonu 1, pozicija 125. ATG kodon, koji kodira metionin , stoga postaje AGG, što predstavlja aminokiselinu arginin , koja se zatim nalazi na poziciji 42 α-galaktozidaze A nakon translacije. To je p.Met42Arg mutacija.
C Ovaj 63-godišnji pacijent ima GT transverziju u eksonu 6, pozicija 982. Kao rezultat razmene nukleotida, GGG kodon postaje TGG, a glicin postaje triptofan , koji se zatim nalazi na poziciji 328 α-galaktozidaze A. Mutacija je stoga označena kao p.Gli328Trp.

Fabrijeva bolest je bolest zasnovana na genetskom defektu (mutaciji) u ženskom polnom hromozomu , H-hromozomu. Svaki otac pogođen bolešću prenosi bolest na sve svoje ćerke, dok svi njegovi sinovi ostaju zdravi. Ako majka nosi mutirani gen, njena deca – bez obzira na pol – imaju 50% rizik da naslede bolest. Mutacija utiče na GLA gen, koji se nalazi na dugom kraku Ks hromozoma na lokusu gena k22.1.[15]
Genski proizvod je homodimer i, kao i svi lizozomalni enzimi, obezbeđen je sa ostatkom manoza-6-fosfata kotranslaciono, odnosno tokom translacije mRNK u sekvencu aminokiselina . Neke od fosforilisanih molekula α-galaktozidaze A luče ćelije i preuzimaju ih druge ćelije preko receptora manoza-6-fosfata vezanog za membranu endocitozoma. Ponovno preuzimanje fosforilisane α-galaktozidaze A preko manoza-6-fosfatnog receptora je osnova za terapiju zamene enzima.[16]
Posebnosti nasleđa vezanog za H uredi
Zbog H-hromozomskog nasleđa, bolest se različito manifestuje kod muškaraca i žena. Muški pacijenti se nazivaju hemizigoti , a žene heterozigotni nosioci. Pretpostavljalo se da samo muškarci mogu da razviju Fabrijevu bolest i da su heterozigotne žene samo nosioci. Ovo je slučaj sa velikom većinom drugih naslednih bolesti povezanih sa H- kao što su hemofilija ili Dišenova mišićna distrofija. Sada je poznato da žene koje su heterozigote za ovu karakteristiku takođe mogu razviti Fabrijevu bolest. Neki autori stoga preporučuju izbegavanje termina H-vezano recesivno , jer je to pogrešno. Umesto toga, preporučuje se terminologija H-vezano nasleđivanje.[17][18][19]
Kod heterozigotnih pacijenata, jedan nemutirani i jedan mutirani H hromozom prisutni su u svakoj ćeliji tela sa DNK sa jezgrom. Inaktivacija H inaktivira jedan od dva H hromozoma u svakoj ćeliji. Ova inaktivacija se dešava nezavisno u svakoj ćeliji i po principu slučajnosti (mozaik ). Prema tome, statistički gledano, 50% ćelija će proizvoditi α-galaktozidazu A sa malo ili bez aktivnosti, u zavisnosti od vrste mutacije. Ostalih 50% ćelija proizvodi α-galaktozidazu A sa normalnom aktivnošću („zdrave ćelije“). Deo aktivne α-galaktozidaze A proizvode „mutantne ćelije“ sa aktiviranim H hromozomom, na kojima je mesto defektnog GLA gena, kao što je prethodno opisano, zauzeto endocitozom. Iako je ovaj prenos enzima dovoljan da spreči imuni sistem da eliminiše mutirane ćelije , on je prenizak da bi se kompenzovao defekt gena kako bi se sprečilo nakupljanje globotriaozilceramida.[20] U poređenju sa drugim lizozomalnim enzimima, unos α-galaktozidaze A prenosom enzima je relativno nizak.[21]
Sa H-inaktivacijom se može objasniti da u proseku kod heterozigotnih žena bolest postaje simptomatska mnogo kasnije i manje je izražena nego kod muškaraca. Međutim, to nije dovoljan model za razumevanje širokog spektra različitih manifestacija bolesti kod žena. Na primer, oko 10% pacijenata zahteva terapiju zamene bubrega tokom progresije bolesti, što odgovara „klasičnom fenotipu“ kod muškaraca. Druge heterozigotne žene, s druge strane, ostaju uglavnom bez simptoma. Razlog za to još nije u potpunosti razjašnjen.[11]
Jedna hipoteza je da nepravilnosti u inaktivaciji H hromozoma igraju važnu ulogu u opsegu varijacija heterozigotnog fenotipa. Ovo se naziva „iskrivljena“ H inaktivacija, u kojoj je statistički očekivani odnos 50:50 između „mutantnih“ i „zdravih“ ćelija jasno pomeren. Ovaj pomak nije uzrokovan prednošću rasta mutantnih ćelija. Indikacija iskrivljene H inaktivacije su heterozigotni pacijenti sa fenotipom kod kojih je Fabrijeva bolest u potpunosti razvijena, pošto je mutirani H hromozom aktiviran u preko 95% ćelija. Otprilike jedan od 200 pacijenata sa Fabrijevom bolešću ima ovaj fenotip. Još jedan pokazatelj iskrivljene H-inaktivacije je ženski heterozigotni par monozigotnih blizanaca , gde je jedan od blizanaca asimptomatski, a drugi je klinički relevantan.[22]
U studiji sa 28 pacijenata, kod većine ispitanika je pronađena iskrivljena inaktivacija H u leukocitima otkriveno, ali nije bilo povezano sa kliničkom manifestacijom bolesti ili sa zaostalom aktivnošću enzima. Autori stoga ne vide nikakvu vezu između fenotipa i iskrivljene H aktivacije.[23]
Mutacione varijante uredi
Defekti GLA gena uzrokovani mutacijama su veoma heterogeni. Do sada je zabeleženo preko 500 različitih mutacija. To uključuje smislene i besmislene tačkaste mutacije, mutacije spajanja, male delecije i umetanja, i velike delecije.[11] Tačkaste mutacije su najčešće (oko 71%), praćene malim delecijama i insercijama koje utiču na manje od 60 nukleotida (oko 27%) i velikim delecijama koje utiču na jedan ili više egzona (oko 2%).[24]
U većini slučajeva, mutacija dovodi do potpunog gubitka aktivnosti enzima.[25]
Neke mutacije koje dovode do promena α-galaktozidaze A i koje su dovoljno udaljene od aktivnog mesta enzima dovode samo do malih strukturnih promena u enzimu, tako da je određena rezidualna aktivnost enzima i dalje prisutna. Takve mutacije kao što su p.Met72Val, p.Gln279Glu ili p.Met296Ile karakteriše blagi fenotip bolesti.[24]
Na GLA genu nema izražene žarišne tačke, to je područje posebno podložno mutacijama. Upadljive su česte rearanžmane DNK u egzonu 7, koji očigledno ima povećanu podložnost preuređivanju.[24]
Patologija uredi
Fabrijeva bolest spada u grupu lizozomalnih bolesti skladištenja, koja obuhvata najmanje 50 članova, i tu u podgrupu sfingolipidoza . Bolest je zasnovana na nedostatku lizozomalnog enzima α-galaktozidaze A. Zbog ovog nedostatka, određeni metabolički proizvodi kao što je globotriaozilceramid (Gb3, Gl3, ranije nazivan i ceramid triheksozid) akumuliraju se u endotelnim ćelijama različitih sistema organa. Smanjena aktivnost α-galaktozidaze u suštini dovodi do akumulacije globotriaozilceramida. Pored toga, akumuliraju se digalaktozailceramid (posebno u bubrezima) i globotriaozilšingozin (lizo-Gb3, lizo-Gl3).[26] Ovi sfingolipidi su važne komponente ćelijske membrane .
Tačne veze između smanjene ili čak potpuno nestale aktivnosti α-galaktozidaze A i patoloških procesa u zahvaćenim organima – koji na kraju dovode do Fabrijeve bolesti – još uvek nisu dovoljno razjašnjene. Raznovrsnost zahvaćenih organa sugeriše da sekundarni biohemijski mehanizmi koji uključuju sfingolipide određuju tok bolesti.[27]
Simptomi opisani u sledećem poglavlju, kao što je progresivna hronična bubrežna insuficijencija , u mnogim publikacijama se pripisuju akumulaciji globotriaozilceramida u lizozomu endotelnih ćelija. Međutim, brojni klinički efekti, posebno u terapiji zamene enzima za Fabrijevu bolest, ne odgovaraju ovom očigledno pojednostavljenom modelu. Na primer, kod nekih pacijenata se mogu primetiti progresivne komplikacije , što sugeriše da ne postoji direktna korelacija između Gb3 i kliničkih manifestacija Fabrijeve bolesti.[28] Zapažanje da je veliki procenat žena GLA- Nosioci mutacije razvijaju simptome slične onima kod hemizigotnih pacijenata, iako ovi pacijenti imaju značajan nivo cirkulišućeg enzima. Pored toga, akumulacija Gb3 u lizozomu hemizigotnih pacijenata počinje u ranom detinjstvu ili pre rođenja, mnogo pre nego što se razviju klinički relevantni simptomi. Takođe ne postoji korelacija između stepena bolesti i nivoa Gb3 u plazmi ili urinu ni kod hemizigotnih ni kod heterozigotnih pacijenata.[29]
Pošto se bolest ne manifestuje u detinjstvu čak ni kod pacijenata bez ikakve aktivnosti α-galaktozidaze A, pretpostavlja se da akumulacija Gb3 nije direktan uzrok Fabrijeve bolesti.
Trenutno se veruje da je globotriaozilsfingozin – metabolit globotriaozilceramida – na kraju uzrok patološkog oštećenja kod Fabrijeve bolesti. Barem u oštećenju glomerula koje dovodi do bubrežne insuficijencije kod Fabrijeve bolesti, lizo-Gb3 igra ključnu ulogu. Liso-Gb3 oslobađa TGF-β1 i inhibitor makrofaga CD74 . Patomehanizam koji sledi podseća na dijabetesnu nefropatiju.[30]
Klinička slika uredi


Gradijentni oblici i ozbiljnost uredi
Pravi se razlika između dva tipa pacijenata i progresivnih oblika Fabrijeve bolesti: „klasičnih“ hemizigotnih pacijenata kod kojih α-galaktozidaza A nema nikakvu aktivnost, i „atipičnih“ heterozigotnih pacijenata kod kojih enzim još uvek ima rezidualnu aktivnost. Klasičan tok bolesti manifestuje se u ranom pojavljivanju simptoma, najčešće u više organa. Nasuprot tome, simptomi se kod atipičnih heterozigotnih pacijenata javljaju mnogo kasnije. Pored toga, u takvim slučajevima bolest može biti lokalizovana, na primer na srčanom mišiću . Muški pacijenti razvijaju tipične simptome Fabrijeve bolesti od detinjstva. Kod žena to često nije slučaj do 40. do 50. godine. Zbog preostale aktivnosti α-galaktozidaze A, simptomi su često manje izraženi.
Simptomi su složeni i mogu se razlikovati od osobe do osobe. Rani simptomi su od velike važnosti za dijagnozu. Većina kasnih simptoma, s druge strane, određuje mortalitet (stopu smrtnosti) pacijenata.
Indeks ozbiljnosti je broj koji određuje težinu bolesti. Za pacijente muške Fabrijeve bolesti, penetrantnost je ocenjena na 100%, a ozbiljnost na 84%. Za pacijentkinje, vrednosti penetracije su 70%, a težine 4%.
Kvalitet života uredi
Simptomi bolesti uzrokuju nizak kvalitet života, posebno kod muških pacijenata. Uporediv je sa onim kod pacijenata sa AIDS-om. Kvalitet života pacijenata sa Fabrijevom bolešću je na sličnom nivou kao kod pacijenata sa multiplom sklerozom ili reumatoidnim artritisom .
Fabrijeva bolest ima značajan negativan uticaj na psihosocijalno okruženje obolelih. Više od polovine muških pacijenata je neoženjeno. Veliki procenat je nezaposlenih. Depresija je izuzetno česta kod pacijenata sa Fabrijevom bolešću. Oni su nedovoljno dijagnostikovani ili se ne leče i značajno smanjuju kvalitet života pacijenata. Prema jednoj studiji, 46% pacijenata ima depresiju, a 28% ima dovoljno tešku da bude klinički relevantna. Rezultati iznad 26 se postižu na Hamiltonovoj skali . Za razliku od normalne populacije, udeo muškaraca sa velikom depresijom (36%) je veći nego kod žena (22%).
Nekoliko studija preporučuje psihijatrijsku i neuropsihološku evaluaciju pacijenata sa Fabrijevom bolešću. Detaljno, pacijenti se žale na fizičku nelagodu, tugu i psihičku patnju . Fizički simptomi se povećavaju pod stresom. Psihološki testovi pokazuju natprosečne poremećaje ponašanja, nepoverenje, defanzivnost, emocionalna previranja i osećaj izolovanosti. Rezultati ovih testova su uglavnom isti kao i kod pacijenata sa bolom.
Rani znaci i simptomi uredi


Bol uredi
Jedan od prvih simptoma klasičnog oblika Fabrijeve bolesti je bol u rukama i stopalima, akralima . Ove akroparestezije se pojavljuju već u detinjstvu. Oni su uzrokovani oštećenjem tankih nervnih vlakana ( neuropatija malih vlakana ) autonomnog i perifernog somatskog nervnog sistema . Oko 60 do 80% dečaka i devojčica sa klasičnom formom je pogođeno ovim bolom.
Pacijenti opisuju dve vrste bola: periodično ponavljajući paroksizmalni napadi bola, koji se nazivaju i „Fabrijeve krize“, sa pekućim bolom koji se širi iz šaka i stopala u druge delove tela, i hronični bol, koji odgovara parestezijama pečenja i peckanja. Fabrijeve krize mogu izazvati groznica, vežba, stres, iscrpljenost i brze promene temperature. Ovi simptomi se ponekad pogrešno tumače kao reumatske tegobe, Rejnoov sindrom , sistemski eritematozni lupus i, pre svega, bolovi u rastu .
Bol obično nestaje u odraslom dobu. Javljaju se ranije i češće kod dečaka nego kod devojčica. Za dečake, u proseku, sa sedam godina, za devojčice sa devet godina. Bol ima značajan negativan uticaj na kvalitet života pacijenata.
Gastrointestinalni poremećaji uredi
Nelagodnost u digestivnom traktu je još jedan čest, uglavnom potcenjen, rani simptom Fabrijeve bolesti. Ove bolesti obično traju u odraslom dobu. Pacijenti se žale na bol u stomaku , obično nakon jela, dijareju , mučninu i povraćanje . Ovo zauzvrat može biti uzrok anoreksije (gubitak apetita). Ovi gastrointestinalni simptomi su verovatno uzrokovani depozitima Gb3 u autonomnim ganglijama (ganglia autonomica) creva i mezenteričnim krvnim sudovima.
Anhidroza uredi
Mnogi pacijenti sa Fabrijevom bolešću ne mogu da se znoje ( anhidroza ) ili to rade samo u znatno smanjenoj meri (hipohidroza). Vrednosti impedanse kože su stoga relativno visoke. Anhidroza/hipohidroza može dovesti do netolerancije na toplotu i značajnih ograničenja u sportskim aktivnostima kod obolelih. U studiji na 714 pacijenata sa Fabrijevom bolešću, anhidroza je dijagnostikovana kod 53% muškaraca i 28% žena. Uzrok smanjene sposobnosti znojenja su akumulacije lipida unutar neurona autonomnog nervnog sistema.
Angiokeratomi uredi
Najlakše prepoznatljivi rani simptom Fabrijeve bolesti je angiokeratom. Ovo su benigne crveno-ljubičaste lezije kože sa blagim izbočinama. Obično se formiraju na zadnjici, preponi, pupku i butini. Povremeno su zahvaćene i sluzokože, na primer u ustima.[32]
U većini slučajeva, angiokeratomi su mali površinski angiomi koji su rezultat oštećenja vaskularnog endotela kože povezanih sa vazodilatacijom u koži. Sa godinama se povećavaju u broju i veličini i mogu se javiti pojedinačno ili u grupama.[33]
-
 Histološka priprema biopsiranog uzorka kože pacijenta sa Fabrijevom bolešću. Na svetlosnoj mikroskopskoj slici, tipične lezije kože se mogu videti kao mali površinski angiomi.
Histološka priprema biopsiranog uzorka kože pacijenta sa Fabrijevom bolešću. Na svetlosnoj mikroskopskoj slici, tipične lezije kože se mogu videti kao mali površinski angiomi. -
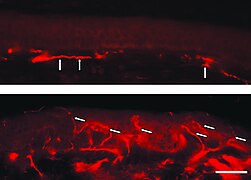 Fluorescentne slike smrznutog dela kože pacijenta sa Fabrijevom bolešću. Nedostatak intraepidermalnih nervnih vlakana i prisustvo vlakana koja pripadaju subepidermalnom nervnom pleksusu (strelice) su upadljivi. Donji uzorak kože, s druge strane, dolazi sa leđa pacijenta. Ovde je upadljiva gusta inervacija epidermisa (strelice).
Fluorescentne slike smrznutog dela kože pacijenta sa Fabrijevom bolešću. Nedostatak intraepidermalnih nervnih vlakana i prisustvo vlakana koja pripadaju subepidermalnom nervnom pleksusu (strelice) su upadljivi. Donji uzorak kože, s druge strane, dolazi sa leđa pacijenta. Ovde je upadljiva gusta inervacija epidermisa (strelice). -
 Crtež nakon posmatranja pod mikroskopom karminskog bojenja epidermisa sa angiokeratomom, akvarel je iz članka Johanesa Fabrija iz 1898. godine
Crtež nakon posmatranja pod mikroskopom karminskog bojenja epidermisa sa angiokeratomom, akvarel je iz članka Johanesa Fabrija iz 1898. godine



Vrtložna keratopatija uredi

Karakteristične zamućenosti rožnjače su najčešći rani simptom Fabrijeve bolesti. Mogu se sa sigurnošću dijagnostikovati pomoću prorezne lampe i javljaju se kod skoro svih hemizigotnih pacijenata. Ovaj oblik zamućenja rožnjače se naziva verticillata rožnjače ili vrtložna keratopatija . Dvostrano je i ima karakterističan uzorak krem boje. Oblačnost ne utiče na oštrinu vida. Neki lekovi, kao što su amiodaron i hlorokin , takođe izazivaju vrtložne keratopatije pri produženoj upotrebi.
Kasni simptomi uredi

Oštećenje bubrega uredi
Kumulativni procenat pacijenata sa Fabrijevom bolešću koji imaju proteinuriju (žuta) i hronična bubrežna insuficijencija (narandžasta). Crvena kriva je stopa smrtnosti. Kao i većina simptoma Fabrijeve bolesti, oštećenje bubrega je progresivno. Završava se terminalnom insuficijencijom bubrega i uzrokuje značajno skraćen životni vek. U klasičnoj kliničkoj slici Fabrijeve bolesti, akumulacije Gb3 u endotelnim ćelijama glomerula , u mezangijalnim ćelijama , u podocitima i u ćelijama intersticijuma dovode do oštećenja bubrega. Ove ćelije su diferencirane epitelne ćelije . Takođe u epitelu Henleove petljeAkumulacije glikosfingolipida nalaze se u bubrežnim arteriolama i distalnim tubulima i u endotelijumu i ćelijama glatkih mišića arteriola . Naslage Gb3 u citoplazmi mogu se jasno videti u transmisionom elektronskom mikroskopu (TEM). Oni su u obliku mijelinskih struktura i naslanjaju se na ćelijsko jezgro . Sa povećanjem akumulacije Gb3, mezangijum se širi, što dovodi do segmentne ili globalne glomeruloskleroze sa zadebljanjem bazalnih membrana . Kao mogući mehanizmi razmatraju se mikrovaskularne lezije i oštećenja podocita, koji su važni za rad filtera, i epitelnih ćelija tubula.
U klasičnom toku bolesti, oštećenje bubrega počinje u drugoj do trećoj deceniji života . Prvo, može se primetiti mikroalbuminurija , što predstavlja izlučivanje malih količina proteina albumina u urinu, koja se razvija u proteinuriju (izlučivanje veće količine proteina urinom). Kurs je sličan dijabetičkoj nefropatiji i direktno doprinosi progresiji Fabri nefropatije. Proteinurija postaje teža sa godinama. Izostenurija se razvija kako bolest napreduje, odnosno bubrezi potpuno gube sposobnost koncentracije ili razblaživanja. Ovo je praćeno promenama tubularne reapsorpcije , sekrecije i izlučivanja .
U početku, oštećenje bubrega je maskirano glomerularnom hiperfiltracijom . Međutim, kada je kritičan broj nefrona oštećen, bubrežna funkcija progresivno opada. Brzina glomerularne filtracije , mera filtracionog kapaciteta bubrega, smanjuje se za oko 12 ml/min godišnje ako se ne leči. U trećoj do petoj deceniji života, funkcija bubrega se postepeno pogoršava i javlja se bubrežna azotemija – ovo je abnormalno povećanje azotnih metabolita kao što su urea i kreatinin u krvi. U ovoj fazi, fibroza , skleroza i tubularna atrofija dominiraju Fabrijevom nefropatijom i na kraju dovode do završnog stadijuma bubrežne bolesti, koja se javlja kod muških pacijenata u četvrtoj do petoj deceniji. Oko 17% svih muškaraca i 1% svih pacijenata obolelih od Fabrijeve bolesti razvije završnu bubrežnu insuficijenciju i zahteva dijalizu. Polovina pacijenata je mlađa od 53 godine. Više od polovine pacijenata sa Fabrijevom bolešću razvija nefropatiju tokom bolesti . Završna bubrežna bolest je glavni faktor uMorbiditet i mortalitet . Bez terapije zamene bubrega, uremija (trovanje urinom) će neizbežno dovesti do smrti.
- Uzorci tkiva iz bubrega pacijenata sa Fabrijevom bolešću
-
 Ovaj svetlosni mikrograf pokazuje akumulaciju Gb3 u glomerularnoj endoteliji, mezangijalnim ćelijama, intersticijskim ćelijama i podocitima.
Ovaj svetlosni mikrograf pokazuje akumulaciju Gb3 u glomerularnoj endoteliji, mezangijalnim ćelijama, intersticijskim ćelijama i podocitima. -
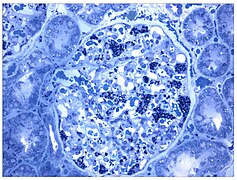 Takođe svetlosni mikrograf. Povećana akumulacija Gb3 u podocitima vizuelizovana je ljubičastom bojom.
Takođe svetlosni mikrograf. Povećana akumulacija Gb3 u podocitima vizuelizovana je ljubičastom bojom. -
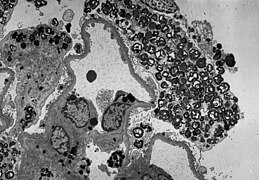 TEM slika pokazuje masivnu elektron-gustu (= crnu) akumulaciju glikosfingolipida u lizozomu podocita
TEM slika pokazuje masivnu elektron-gustu (= crnu) akumulaciju glikosfingolipida u lizozomu podocita -
 Takođe TEM slika. Prikazuje inkluzije glikosfingolipida različitih oblika i veličina u ćelijama distalnog tubula.
Takođe TEM slika. Prikazuje inkluzije glikosfingolipida različitih oblika i veličina u ćelijama distalnog tubula. -
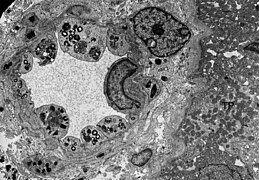 TEM slika endotelije i glatkih mišićnih ćelija bubrežne arteriole sa inkluzijama glikosfingolipida
TEM slika endotelije i glatkih mišićnih ćelija bubrežne arteriole sa inkluzijama glikosfingolipida





Oštećenje srca uredi

Oko 40 do 60% ljudi sa Fabrijevom bolešću ima srčane simptome kao što su hipertrofija leve komore (zadebljanje leve komore srca), abnormalni srčani ritam (aritmija), angina pektoris (bol u grudima sličan napadu) i dispneja (otežano disanje).[34][35] Srčanu aritmiju i poremećenu varijabilnost srčane frekvencije uzrokuju sinusni čvor, provodni sistem i neravnoteža između tonusa simpatikusa i parasimpatikusa. Dijastolna disfunkcija i hipertrofija leve komore su važni simptomi Fabrijeve bolesti. Ovi simptomi su generalno ozbiljniji za muškarce nego za žene. Ishemija miokarda (poremećaji cirkulacije srčanog mišića) je rezultat lošeg koronarnog protoka krvi.[36]
U starosti se razvija progresivna fibroza miokarda, koja je i reverzibilno intersticijalna i ireverzibilno ožiljna (zamena fibroze).[37] U skoro svim slučajevima, ireverzibilne ožiljne fibroze formiraju se prvo u zadnjem bočnom srčanom zidu iu srednjem miokardu. Kod pacijenata u krajnjem stadijumu, transmuralna (obuhvata celu debljinu zidnog sloja srca) ožiljna fibroza postepeno smanjuje srčanu funkciju do tačke kongestivne srčane insuficijencije.[38] Maligne aritmije su odgovorne za većinu srčanih smrti kod pacijenata sa Fabrijevom bolešću.[34][11][39]
Strukturne promene leve komore u srcu su uobičajene kod pacijenata sa Fabrijevom bolešću. Uglavnom koncentrične hipertrofije,[40] mogu se učiniti vidljivim ehokardiografijom (ultrazvukom srca) ili magnetnom rezonancom srca (MRT). Pošto zadnji zid leve komore srca postaje sve tanji i tanji sa starenjem zbog zamene fibroze, merenje debljine septuma - to je debljina pregradnog zida između leve i desne polovine srca - je posebno važno. Bez obzira na strukturne promene, sistola, faza u kojoj se krv istiskuje iz leve i desne komore, izgleda da je u velikoj meri očuvana kada se meri konvencionalnim metodama.[41] Kardiomiopatiju uzrokovanu Fabrijevom bolešću karakteriše smanjena kontrakcija i opuštanje srčanog mišića. Dopler tkiva (i snimanje brzine tkiva i slika brzine naprezanja) može kvantifikovati funkciju srčanog mišića.[11] Ovom metodom, kardiomiopatija se može dijagnostikovati pre nego što se razvije hipertrofija leve komore.[42]
- Ehokardiogrami pacijenata sa Fabrijevom bolešću
-
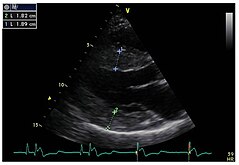 Parasternalna duga osa: Hipertrofija leve komore sa povećanom debljinom septuma je jasno vidljiva.
Parasternalna duga osa: Hipertrofija leve komore sa povećanom debljinom septuma je jasno vidljiva. -
 Parasternalna kratka osa: slika takođe pokazuje hipertrofiju leve komore.
Parasternalna kratka osa: slika takođe pokazuje hipertrofiju leve komore. -
 Dopler ehokardiografija mitralnog prstena (mitralnog prstena) sa skoro normalnom sistolnom funkcijom
Dopler ehokardiografija mitralnog prstena (mitralnog prstena) sa skoro normalnom sistolnom funkcijom




Kod mnogih bolesnika sa Fabrijevom bolešću, desna komora je takođe hipertrofična (hipertrofija desne komore). Komora je normalne veličine i sistola je takođe normalna; dijastolna funkcija je značajno smanjena. Dve trećine pacijenata sa hipertrofijom desne komore takođe pokazuju simptome hipertrofija desne komore.[43]
Hipertrofija desne komore je verovatan razlog zašto pacijenti sa dobrom funkcijom srca leve komore imaju nisku fizičku izdržljivost i pate od organomegalije (uvećanje organa) i limfedema.[11][44]
Zbog oštećenja srčane funkcije, elektrokardiogrami (EKG) odraslih pacijenata obolelih od Fabrijeve bolesti sa klasičnim oblikom bolesti pokazuju karakteristične promene.[11]
Cerebrovaskularno oštećenje uredi
Rani simptomi perifernih neuropatija, koji se obično javljaju u adolescenciji, često su praćeni u odraslom dobu cerebrovaskularnim oboljenjima i autonomnim disfunkcijama (bolesti ili funkcionalni poremećaji autonomnog nervnog sistema). Neke od najnepovoljnijih neuroloških karakteristika Fabrijeve bolesti su uzrokovane cerebralnim višestrukim (multifokalnim) poremećajima cirkulacije u malim krvnim sudovima.[45] Cerebrovaskularne promene mogu dovesti do različitih znakova i simptoma. Spektar se kreće od glavobolje i vrtoglavice, preko prolaznih ishemijskih napada i ishemijskih moždanih udara,[46] do vaskularne demencije.[11][47]
Prevalencija cerebralnog infarkta je oko 6,9% kod muškaraca i 4,3% kod žena. Značajno je veći nego u opštoj populaciji. Srednja starost kod prvog moždanog udara je oko 39 godina za muškarce sa Fabrijevom bolešću i 46 za žene. Moždani infarkt je neretko prva manifestacija Fabrijeve bolesti.[45] U većini slučajeva, infarkt mozga izazivaju mali krvni sudovi. Pored toga, dolihoektazije (sin. dilatativne arteriopatije, proširenje arterija) vertebrobazilarne cirkulacije su takođe opisane kao okidači.[11] Formiranje tromba može biti podstaknuto povećanom adhezijom neutrofilnih granulocita i monocita na endotel ili lokalnim povećanim protokom krvi (hiperperfuzija).[11][48] Nivo enzima mijeloperoksidaze u serumu je biomarker za rizik od vaskulopatije kod muškaraca sa Fabrijevom bolešću.[11]
-
 MRI aksijalnog mozga 27-godišnjeg pacijenta sa Fabrijevom bolešću sa ishemijskim moždanim udarom koji pokazuje moždani udar u levoj hemisferi malog mozga. Pacijent nije imao drugih simptoma bolesti
MRI aksijalnog mozga 27-godišnjeg pacijenta sa Fabrijevom bolešću sa ishemijskim moždanim udarom koji pokazuje moždani udar u levoj hemisferi malog mozga. Pacijent nije imao drugih simptoma bolesti -
 Hiperintenzitet bele materije , lakunarni cerebralni infarkt i mikrokrvarenje.
Hiperintenzitet bele materije , lakunarni cerebralni infarkt i mikrokrvarenje. -
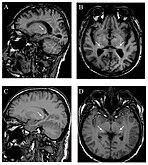 T1 - ponderisani sagitalni (A) i aksijalni (B) MRI pokazuju simetrično visok signal u talamusu (tzv. pulvinarski znak ) 66-godišnjeg pacijenta. (C) i (D), takođe T 1 -ponderisani, pokazuju pulvinarski znak kod 42-godišnjeg pacijenta.
T1 - ponderisani sagitalni (A) i aksijalni (B) MRI pokazuju simetrično visok signal u talamusu (tzv. pulvinarski znak ) 66-godišnjeg pacijenta. (C) i (D), takođe T 1 -ponderisani, pokazuju pulvinarski znak kod 42-godišnjeg pacijenta. -
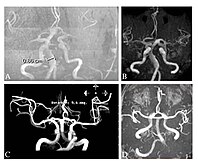 Vremenska magnetna rezonantna angiografija četiri pacijenta sa Fabrijevom bolešću pokazuje proširene (ekstatične) krvne sudove (dolihoektazija vertebrobazilarne cirkulacije).
Vremenska magnetna rezonantna angiografija četiri pacijenta sa Fabrijevom bolešću pokazuje proširene (ekstatične) krvne sudove (dolihoektazija vertebrobazilarne cirkulacije).




Ostali kasniji efekati bolesti uredi


Oštećenje bubrega, srca i mozga čini najveći deo smrtnosti od Fabrijeve bolesti. Druge kasne posledice su klinički relevantne, ali malo ili nimalo doprinose mortalitetu bolesti. Na primer, oštećenja organa sluha i ravnoteže su široko rasprostranjena. 80% muških i 77% ženskih pacijenata pokazuje progresivni gubitak ravnoteže.[49] Funkcija vestibularnog sistema može se proveriti pomoću impulsnog testa glave. Kod hemizigotnih pacijenata sa klasičnim tokom bolesti, progresivni gubitak sluha i iznenadna gluvoća su izuzetno česti.[50] Akumulacija α-galaktozidaze A takođe može dovesti do tinitusa i vrtoglavice.[51]

Disajni putevi su takođe pogođeni bolešću kod mnogih pacijenata sa Fabrijevom bolešću. Otežano disanje (dispneja), hronični kašalj, piskanje i piskanje su uobičajeni kod oba pola.[52] Prema jednoj studiji, 61% muškaraca i 26% žena ima opstrukcije disajnih puteva.[11]
Promene na skeletu, koje suštinski utiču na gustinu kostiju, takođe su čest kasni simptom Fabrijeve bolesti. U jednoj studiji, kod 88% pacijenata prosečne starosti od 31 godine i klasičnog toka bolesti dijagnostikovana je ili osteopenija ili uznapredovali stadijum, osteoporoza, korišćenjem dualne rendgenske apsorpciometrije (DKSA).[53] U sledećoj većoj studiji, osteopenija je pronađena kod približno 50% pacijenata sa Fabrijevom bolešću. Smanjena gustina kostiju može dovesti do spontanih preloma.[54][55]
Dijagnoza uredi
Važnost rane dijagnoze uredi
Dijagnostikovanje Fabrijeve bolesti što je ranije moguće je važno iz više razloga. S jedne strane, od 2001. godine postoji mogućnost da se bolest leči uzročno. Kvalitet života obolelih može se značajno poboljšati, a oštećenje organa barem smanjiti ili odložiti. S druge strane, genetska predispozicija za bolest može se prepoznati kod članova porodice pre nego što se pojave prvi simptomi. U takvim slučajevima moguće je praćenje razvoja bolesti i rana terapija pre nego što bolest postane simptomatska.[56]
Pogrešna dijagnoza uredi
Zbog retkosti bolesti, većina pedijatara i internista pogrešno postavlja dijagnozu Fabrijeve bolesti i netačno je leči. Studija iz 2010. analizirala je istoriju bolesti 45 pacijenata sa Fabrijevom bolešću. Većina pacijenata se u mladosti žalila na neuropatski bol kao prvi simptom bolesti - u većini slučajeva pogrešno je dijagnostifikovan kao "reumatska groznica". Sedam pacijenata je godinama lečeno penicilinom. Deset pacijenata sa bolovima u stomaku dijagnostikovano je trovanje hranom ili "nespecifični bol". Početnom simptomu anhidroze nije bilo moguće pripisati uzrok i angiokeratomi su interpretirani kao petehije. U proseku je bilo potrebno 19,7 godina da se postavi tačna dijagnoza Fabrijeve bolesti.[57] U prethodnoj studiji na 366 pacijenata, ova vremenska razlika je bila 13,7 godina za muškarce i 16,3 godine za žene.[58] Britanska studija iz 2001. godine pronašla je srednju starost od 22 godine za muške pacijente u vreme inicijalne dijagnoze, koja je postavljena u prosečnom intervalu od 8 godina nakon prvih simptoma.[59]
U dugom periodu između prvih simptoma i tačne dijagnoze, mnogi pacijenti moraju da izdrže dugu i frustrirajuću odiseju od lekara do lekara.[60] U većini slučajeva tačnu dijagnozu postavlja slučajno oftalmolog preko verticillata rožnjače (vorteks keratopatija) ili dermatolog preko angiokeratoma.[59]
Tačna dijagnoza uredi
U slučaju klasičnog oblika Fabrijeve bolesti, klinička slika može značajno doprineti ranoj ispravnoj dijagnozi; posebno angiokeratomi i vorteks keratopatija. Određivanje aktivnosti α-galaktozidaze A iz plazme ili iz leukocita pomoću enzimskog testa daje dijagnostičku sigurnost kod pacijenata muškog pola.[61] Određivanje iz plazme ponekad može dovesti do pogrešne dijagnoze Fabrijeve bolesti, zbog čega se preporučuje da se nalaz proveri određivanjem aktivnosti leukocita.[62] Kod ženskih pacijenata ova metoda je često nedovoljna. Ne uspeva kod više od 30% pacijenata sa Fabrijevom bolešću jer je rezidualna aktivnost enzima previsoka za test. [63] Stoga, svim pacijentima za koje se sumnja da imaju Fabrijevu bolest treba dijagnostikovati genotipizaciju.[18]
Gen je sekvencioniran i uparen sa poznatim GLA mutacijama. Pored toga, korisno je određivanje biomarkera Liso-Gb3. Ako je aktivnost enzima nejasna, vrednost Liso-Gb3 veća od 1,3 nmol/l može ukazivati na Fabrijevu bolest kod ljudi sa nespecifičnim simptomima Fabrijeve bolesti (LVH ili CKD, itd.).[64] Test osušene krvi (DBS) koji se lako može integrisati u svakodnevnu praksu sada je dostupan za merenje aktivnosti enzima i Liso-Gb3, kao i za genetsku analizu: nekoliko kapi krvi stavlja se na karticu osušene krvi. Nakon što se osuše, kartica se šalje poštom u specijalizovanu laboratoriju. Tamo se krv uklanja sa filter kartice i priprema za dalja ispitivanja. Lizo-GL-3 je dobar marker da se sa sigurnošću isključi klasična Fabrijeva bolest. Kod osoba sa neizvesnim GLA značajem, koje pokazuju nespecifične simptome Fabrijeve bolesti (LVH ili CKD, itd.) i nemaju karakteristične, fenotipske ili biohemijske karakteristike klasične Fabrijeve bolesti, treba koristiti > 1,3 nmol/L misliti na Fabrijevu bolest.[64]
U principu, nivoi Gb3 u plazmi i urinu,[29] takođe se mogu koristiti za dijagnozu.[65] Vrednosti urina su pouzdanije od vrednosti u plazmi u pogledu njihove informativne vrednosti kod muških i ženskih pacijenata; međutim, neki pacijenti sa kasnim stadijumom Fabrijeve bolesti ili sa određenim mutacijama (kao što je p.Asn215Ser) imaju normalne koncentracije Gb3 u urinu.[11]
Kobov sindrom se mora razlikovati u diferencijalnoj dijagnozi.
Prenatalna dijagnoza uredi
Dijagnoza Fabrijeve bolesti je prenatalna, odnosno prenatalna. Za ovo se može koristiti biohemijska ili molekularna prenatalna dijagnostika. U prvom slučaju, aktivnost α-galaktozidaze A horionskih resica se može meriti direktno ili u ćelijskoj kulturi. Uklanjanje uzorkovanjem horionskih resica moguće je u desetoj nedelji trudnoće. Dijagnoza je moguća oko 14. nedelje trudnoće iz kultivisanih amnionskih ćelija (ćelija u amnionskoj tečnosti), koje se iz plodove vode uklanjaju amniocentezom. Određivanje genotipa pomoću DNK analize (genski test) je komplikovanije. Genetsko savetovanje se obično obavlja pre prenatalne dijagnoze. Iz etičkih razloga, prenatalna dijagnoza Fabrijeve bolesti, posebno kod ženskih fetusa, veoma je kontroverzna. Sa dostupnošću terapije zamene enzima, ova diskusija se proširila na muške fetuse. Neki autori generalno preporučuju prenatalnu dijagnozu samo za muške fetuse. Pol fetusa se može odrediti iz krvi majke u 9. do 11. nedelji trudnoće.[11]
U principu, preimplantaciona dijagnostika je moguća i već je sprovedena. Do sada (od septembra 2011.) nije bilo publikacija o tome.[66]
Skrining novorođenčeta uredi
Morbus Fabri trenutno nije deo projekcija novorođenčadi u Nemačkoj i Austriji. Pretpostavka je da skrining za određenu urođenu bolest ima smisla samo ako postoji i opcija lečenja za nju. Sa dostupnošću terapije zamene enzima, ova premisa kod Fabrijeve bolesti više ne važi.[67] HPLC-MS se može koristiti za brzu i relativno jeftinu analizu nekoliko lizozomalnih bolesti skladištenja od suvih krvnih mrlja (DBS).[68] Trenutno je u toku nekoliko velikih studija kako bi se potvrdila pouzdanost metode.[68] Lizo-GL-3 plazme se može lako meriti testom suve krvi i nudi efikasan i specifičan biomarker za skrining novorođenčadi za koje se sumnja da imaju Fabrijevu bolest pre nego što se pojave prvi simptomi.[69]
Terapija uredi

Kao i kod svih bolesti lizozomskog skladištenja, razvoj efikasnih metoda lečenja, posebno aktivnih sastojaka, je izuzetno težak, pa tako i za Fabrijevu bolest. S jedne strane, zbog niske incidence, postoji vrlo mali broj pacijenata za sprovođenje kliničkih studija, a sa druge strane, zahtevi u pogledu bezbednosti leka kada se uzimaju tokom dužeg vremenskog perioda su veoma visoki. Lek treba uzimati doživotno, a idealno pre nego što se pojave prvi simptomi, odnosno uglavnom zdravi pacijenti.[70] Zbog retkosti bolesti, tržište za razvijeni lek je izuzetno malo. Visoki troškovi razvoja koji su uobičajeni u farmaceutskoj industriji se tako raspoređuju na mali broj pacijenata, što rezultuje veoma visokim troškovima lečenja po pacijentu.
Do 2001. godine pacijenti sa Fabrijevom bolešću mogli su se lečiti samo simptomatski ili palijativno . Do ove tačke, tretman se u suštini sastojao od izbegavanja stimulansa koji izazivaju bol kao što su stres, fizički napor, toplota, sunčeva svetlost i oštre promene temperature. Za ublažavanje bola korišćene su visoke doze analgetika . Anhidroza je suzbijana povećanjem unosa tečnosti po toplom vremenu i izbegavanjem fizičkog napora. Dijeta sa malo masti i lekovi su korišćeni za ublažavanje gastrointestinalnih simptoma. dijeta za bubregeje propisan za blagu proteinuriju. Antikoagulansi su propisani za sprečavanje moždanog udara. Završna bubrežna insuficijencija je – kao što je i danas slučaj – lečena terapijom zamene bubrega (dijaliza ili transplantacija bubrega).[71]
Terapija zamene enzima uredi
Enzimska supstituciona terapija (ERT) je trenutno (od septembra 2011.) jedina mogućnost za uzročni tretman ( kauzalna terapija ) Fabrijeve bolesti.[72] Za pacijente sa Fabrijevom bolešću u Evropskoj uniji postoje dva leka koji se koriste za lečenje bolesti: agalzidaza alfa i agalzidaza beta. Obe su biotehnološki proizvedene varijante α-galaktozidaze A. Agalsidaza beta je dostupna pacijentima u SAD od aprila 2003. godine. Do danas (od septembra 2011.), agalsidaza alfa nije odobrena u Sjedinjenim Američkim Državama. Osim u 27 zemalja EU, odobren je u ukupno 45 zemalja, uključujući Kanadu, Japan, Brazil i Kinu (od maja 2011).[73]
Oba leka su genetski modifikovana. Dok ćelijska linija humanih fibroblasta proizvodi enzim za agalzidazu alfa, ćelije jajnika kineskog hrčka ( CHO) se koriste za agalzidazu beta.[74] Agalsidaza beta je himerni protein. Dva enzima su identična u svojoj sekvenci aminokiselina, ali se neznatno razlikuju po tipu glikozilacije zbog različitog oblika ekspresije proteina tokom proizvodnje . Glavne razlike su u proporciji sijaličnih kiselinai manoza-6-fosfat. Agalsidaza beta ima veći udeo potpuno sijalirovanih oligosaharida i viši stepen fosforilacije. U in vitro eksperimentima, povećano vezivanje za manoza-6-fosfatni receptor i veće uzimanje u Fabri fibroblastima moglo se utvrditi za agalzidazu beta.[75] Nasuprot tome, nikakva funkcionalna razlika između dva enzima se ne može identifikovati in vivo . Takođe se ne razlikuju u pogledu stepena unakrsne reaktivnosti antitela . Oba enzima se moraju primeniti intravenozno. Enzimi koji bi trebalo da imaju sistemski efekat u osnovi nisu dostupni oralno, jer se u crevima u velikoj meri razlažu na svoje komponente (aminokiseline). Agalsidaza alfa se primenjuje tokom 40 minuta u dozi od 0,2 mg/kg telesne težine svake dve nedelje . Agalsidaza beta se primenjuje istom brzinom.[72] Međutim, doza je 1 mg/kg telesne težine, a trajanje infuzije je u početku četiri sata. Ovo vreme infuzije se onda može smanjiti na 90 minuta ako se dobro podnosi.
Efikasnost uredi
Osip kod 39-godišnjeg pacijenta nakon infuzije α-galaktozidaze A. Pacijent je razvio imunoglobulin E protiv agalzidaze beta. Uprkos primeni oralnih kortikosteroida , paracetamola i hidroksizina , tokom faza infuzije su se razvili opsežni osip na koži i bronhospazam . Funkcija bubrega je nastavila da se pogoršava. Zbog toga je terapija zamene enzima prekinuta. Pacijent ima mutaciju p.Ala121Pro koja se ne može lečiti ispitivanim migalastatom. U vreme prijema, razgovaralo se o kombinaciji imunosupresije i terapije zamene enzima radi daljeg lečenja. Zbog sporog napredovanja Fabrijeve bolesti tokom decenija, kao i zbog njene retkosti, postoji samo nekoliko pouzdanih podataka o dugoročnim uspesima lečenja. U komparativnoj studiji u periodu od 24 meseca, nije pronađena razlika između agalzidaze alfa i agalzidaze beta u pogledu osnovnih merljivih parametara bolesti. Naročito u endotelnim ćelijama, terapija zamene enzima može značajno da smanji akumulaciju Gb3 u lizozomu. Ovo je od velikog značaja za lečenje primarnih faktora morbiditeta (bubrežna insuficijencija, srčana i cerebrovaskularna oboljenja). Međutim, efikasnost terapije kod pacijenata sa uznapredovalim simptomima je prilično skromna. Zbog toga je što ranije moguće lečenje posebno važno za uspeh terapije. U terapiji zamene enzima, degradacija Gb3 zavisi od tipa ćelije. U bubrezima, Gb3 se takođe razgrađuje u mezangijalnim ćelijama glomerula i ćelijama u intersticijumu bubrežnog korteksa, pored vaskularne endotelije. Smanjenje Gb3 u glatkim mišićima arteriola i malih arterija, podocitima i u epitelu distalnog tubula je značajno lošije.
U kliničkim ispitivanjima, primena agalzidaze alfa je smanjila bol i smanjila hipertrofiju leve komore. Stabilizirana je funkcija bubrega, poboljšan sluh i sposobnost znojenja. Sve u svemu, kvalitet života je značajno poboljšan. Kod pacijenata sa hroničnom bubrežnom insuficijencijom, progresija do završnog stadijuma bubrežne insuficijencije može biti odložena. Tokom tretmana agalzidazom beta, detektovano je smanjenje Gb3 u različitim ćelijama. Takođe nema ponovne akumulacije Gb3. Primena agalzidaze beta značajno smanjuje rizik od ozbiljnog kliničkog događaja (npr. infarkt miokarda, završna bubrežna bolest ili smrt).[76]
Neželjeni efekti uredi

Otprilike polovina svih pacijenata doživljava blage do umerene reakcije povezane sa infuzijom, koje dostižu vrhunac između pete i osme infuzije. Pored toga, neki pacijenti pokazuju groznicu i mrzlicu. Ovi neželjeni efekti su kratkotrajni, nisu ozbiljni i mogu se lečiti konzervativno. Posle tri do pet godina lečenja, samo 10 do 20% pacijenata i dalje pokazuje reakcije povezane sa infuzijom, pri čemu se očigledno razvija tolerancija na infuziju. Još uvek nije poznat tačan uzrok reakcija povezanih sa infuzijom.
Postoji sumnja na specifično IgG antitelo-Edukacija o infuziranom enzimu. IgG serokonverzija je otkrivena kod 24% pacijenata koji su primali agalsidazu alfa i 88% pacijenata koji su primali agalsidazu beta. Do formiranja antitela može doći kod pacijenata koji nemaju nikakvu rezidualnu aktivnost α-galaktozidaze. α-galaktozidaza je "nova" i "vanzemaljska" njihovom imunološkom sistemu. Formiranje antitela protiv dva preparata agalzidaze direktno smanjuje njihovu efikasnost.[78] Međutim, studija sa više centara je pokazala da je „prekomerno prskanje“ antitela odgovarajućim količinama enzima rezultiralo kontinuiranim niskim nivoima markera bolesti Liso-Gb3.[79]
Troškovi terapije uredi
Troškovi lečenja u Nemačkoj su oko 250.000 evra po pacijentu godišnje, bez obzira da li se koristili agalzidazu alfa ili beta.[80] Pošto je zamenska terapija enzimima, trenutno jedina uzročna terapijska opcija za Fabrijevu bolst, troškove za to nadoknađuju obavezna zdravstvena osiguravajuća društva u Nemačkoj i ne računaju se u budžet lekarske ordinacije za recept.[81]
Ampula koja sadrži 3,5 mg agalzidaze alfa, za dozu od 0,2 mg po kg telesne težine, košta 1.685 evra u Francuskoj. Za ampulu sa 35 mg agalzidaze beta, sa dozom od 1 mg po kg telesne težine, naplaćuje se 3.370 €. Ovo rezultuje identičnim godišnjim troškovima lečenja po pacijentu, koji u Francuskoj iznose 161.781 € za pacijenta težine 70 kg (2009. godine).[11]
Istovremene terapije uredi
Naročito kod mladih pacijenata, terapija zamene enzima je prilično uspešna u smanjenju neuropatskog bola.[82] Međutim, u mnogim slučajevima je indikovana istovremena terapija bola . karbamazepinom,[83] ako je potrebno u kombinaciji sa pregabalinom,[84] preporučuje se kao lek prvog izbora za lečenje neuropatskog bola kod Fabrijeve bolesti .
U slučaju nepodnošljivih bolnih kriza, mogu se koristiti i opioidi.[85] Lekovi kao što je metoklopramid se koriste za lečenje gastrointestinalnih simptoma preporučuje se za suzbijanje poremećaja kretanja u gornjem delu gastrointestinalnog trakta (poremećaji motiliteta).[86]
Ako postoji povećano izlučivanje proteina u urinu kao indikacija oštećenja bubrega, dodatno lečenje ACE inhibitorima ili AT1 antagonistima , dve srodne klase antihipertenzivnih lekova, može odložiti napredovanje oštećenja bubrega.[87] Ako je srce već oštećeno, ACE inhibitori mogu smanjiti arterijski vaskularni otpor, a time i arterijski krvni pritisak. Ovo takođe smanjuje predopterećenje i naknadno opterećenje srca u slučaju miokardne insuficijencije i povećava minutni volumen srca . Broj otkucaja srca i potreba miokarda za kiseonikom mogu se smanjiti beta-blokatorima. Srčane aritmije se mogu korigovati npr. amiodaronom. Pored toga, još uvek postoje hirurške mere, kao što su implantacija pejsmejkera , koronarnog stenta , veštačkog srčanog zaliska ili premosnice koronarne arterije.[88]
Izvori uredi
- ^ a b v g d Zarate, Yuri A.; Hopkin, Robert J. (2008-10-18). „Fabry's disease”. The Lancet (na jeziku: engleski). 372 (9647): 1427—1435. ISSN 0140-6736. PMID 18940466. S2CID 21137862. doi:10.1016/S0140-6736(08)61589-5.
- ^ Warnock, D. G.; West, M. L. (2006). „Diagnosis and management of kidney involvement in Fabry disease”. Adv Chronic Kidney Dis. 13 (2): 138—147. PMID 16580615. doi:10.1053/j.ackd.2006.01.013.
- ^ a b MacDermot, K. D.; Holmes, A.; Miners, A. H. (1. 11. 2001). „Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females”. Journal of Medical Genetics. 38 (11): 769—775. ISSN 1468-6244. PMC 1734754
 . PMID 11732485. doi:10.1136/jmg.38.11.769.
. PMID 11732485. doi:10.1136/jmg.38.11.769.
- ^ Fabry, H. (2001). „An historical overview of Fabry disease”. Journal of Inherited Metabolic Disease. 24: 3—7. PMID 11758677. S2CID 38947917. doi:10.1023/A:1012443001449.
- ^ Mehta, A.; Beck, M.; Eyskens, F.; Feliciani, C.; Kantola, I.; Ramaswami, U.; Rolfs, A.; Rivera, A.; Waldek, S.; Germain, D. P. (2010). „Fabry disease: A review of current management strategies”. QJM. 103 (9): 641—59. PMID 20660166. doi:10.1093/qjmed/hcq117..
- ^ Desnick, R. J.; Brady, R.; Barranger, J.; Collins, A. J.; Germain, D. P.; Goldman, M.; Grabowski, G.; Packman, S.; Wilcox, W. R. (2003). „Fabry disease, an under-recognized multisystemic disorder: Expert recommendations for diagnosis, management, and enzyme replacement therapy”. Annals of Internal Medicine. 138 (4): 338—46. PMID 12585833. S2CID 12227778. doi:10.7326/0003-4819-138-4-200302180-00014.
- ^ Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R. J. (2006). „High incidence of later-onset Fabry disease revealed by newborn screening”. Am J Hum Genet. 79 (1): 31—40. PMC 1474133 . PMID 16773563. doi:10.1086/504601..
- ^ a b Gold, K. F.; Pastores, G. M.; Botteman, M. F.; Yeh, J. M.; Sweeney, S.; Aliski, W.; Pashos, C. L. (2002). „Quality of life of patients with Fabry disease”. Quality of Life Research : An International Journal of Quality of Life Aspects of Treatment, Care and Rehabilitation. 11 (4): 317—327. PMID 12086117. S2CID 25530468. doi:10.1023/a:1015511908710.
- ^ C. Vetter: Repetitorium: Morbus Fabry. (PDF) In: ZM. Band 101, Nr. 1A, vom 1. Januar 2011, S. 41–45.
- ^ Hwu, W. L.; Chien, Y. H.; Lee, N. C.; Chiang, S. C.; Dobrovolny, R.; Huang, A. C.; Yeh, H. Y.; Chao, M. C.; Lin, S. J.; Kitagawa, T.; Desnick, R. J.; Hsu, L. W. (2009). „Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A)”. Human Mutation. 30 (10): 1397—1405. PMC 2769558 . PMID 19621417. doi:10.1002/humu.21074., 2009, S. 1397–1405
- ^ a b v g d đ e ž z i j k l lj m n Germain, Dominique P. (2010). „Fabry disease”. Orphanet Journal of Rare Diseases. 5: 30. PMC 3009617 . PMID 21092187. doi:10.1186/1750-1172-5-30 .
- ^ Lin, Hsiang-Yu; Chong, Kah-Wai; Hsu, Ju-Hui; Yu, Hsiao-Chi; Shih, Chun-Che; Huang, Cheng-Hung; Lin, Shing-Jong; Chen, Chen-Huan; Chiang, Chuan-Chi; Ho, Huey-Jane; Lee, Pi-Chang; Kao, Chuan-Hong; Cheng, Kang-Hsiang; Hsueh, Chuen; Niu, Dau-Ming (2009). „High Incidence of the Cardiac Variant of Fabry Disease Revealed by Newborn Screening in the Taiwan Chinese Population”. Circulation: Cardiovascular Genetics. 2 (5): 450—456. PMID 20031620. S2CID 10456593. doi:10.1161/CIRCGENETICS.109.862920..
- ^ Gaspar, Paulo; Herrera, Julio; Rodrigues, Daniel; Cerezo, Sebastián; Delgado, Rodrigo; Andrade, Carlos F.; Forascepi, Ramón; MacIas, Juan; Del Pino, Maria D.; Prados, Maria D.; De Alegria, Pilar R.; Torres, Gerardo; Vidau, Pedro; Sá-Miranda, Maria C. (2010). „Frequency of Fabry disease in male and female haemodialysis patients in Spain”. BMC Medical Genetics. 11: 19. PMC 2837018 . PMID 20122163. doi:10.1186/1471-2350-11-19 .
- ^ Nowak, A.; Mechtler, T. P.; Hornemann, T.; Gawinecka, J.; Theswet, E.; Hilz, M. J.; Kasper, D. C. (2018). „Genotype, phenotype and disease severity reflected by serum LysoGb3 levels in patients with Fabry disease” (PDF). Mol Genet Metab. 123 (2): 148—153. PMID 28728877. doi:10.1016/j.ymgme.2017.07.002.
- ^ „OMIM Gene/Loci: 413 – 422 of 718 on Chromosome X”. www.omim.org. Pristupljeno 2022-02-23.
- ^ „Fabry Disease”. The Medical Biochemistry Page (na jeziku: engleski). 2020-05-15. Pristupljeno 2022-02-23.
- ^ Pinto, Louise LC; Vieira, Taiane A.; Giugliani, Roberto; Schwartz, Ida VD (2010). „Expression of the disease on female carriers of X-linked lysosomal disorders: A brief review”. Orphanet Journal of Rare Diseases. 5: 14. PMC 2889886 . PMID 20509947. doi:10.1186/1750-1172-5-14 .
- ^ a b Germain, D.P.; Benistan, K.; Angelova, L. (2010). „X-linked inheritance and its implication in the diagnosis and management of female patients in Fabry disease”. La Revue de Médecine Interne. 31: S209—S213. PMID 21211665. doi:10.1016/S0248-8663(10)70013-8..
- ^ Dobyns, William B.; Filauro, Allison; Tomson, Brett N.; Chan, April S.; Ho, Allen W.; Ting, Nicholas T.; Oosterwijk, Jan C.; Ober, Carole (2004). „Inheritance of most X-linked traits is not dominant or recessive, just X-linked”. American Journal of Medical Genetics. 129A (2): 136—143. PMID 15316978. S2CID 42108591. doi:10.1002/ajmg.a.30123.
- ^ Migeon, Barbara R. (2008). „X Inactivation, Female Mosaicism, and Sex Differences in Renal Diseases”. Journal of the American Society of Nephrology. 19 (11): 2052—2059. PMID 18448583. doi:10.1681/ASN.2008020198.. (Review).
- ^ Kobayashi, M.; Ohashi, T.; Sakuma, M.; Ida, H.; Eto, Y. (2008). „Clinical manifestations and natural history of Japanese heterozygous females with Fabry disease”. Journal of Inherited Metabolic Disease. 31: 483—487. PMID 18202903. S2CID 32234710. doi:10.1007/s10545-007-0740-6..
- ^ Redonnet-Vernhet, I; Ploos van Amstel, J K; Jansen, R P; Wevers, R A; Salvayre, R; Levade, T (1996). „Uneven X inactivation in a female monozygotic twin pair with Fabry disease and discordant expression of a novel mutation in the alpha-galactosidase A gene.”. Journal of Medical Genetics. 33 (8): 682—688. ISSN 0022-2593. PMC 1050704 . PMID 8863162. doi:10.1136/jmg.33.8.682.
- ^ Maier, Esther M.; Osterrieder, Stephanie; Whybra, Catharina; Ries, Markus; Gal, Andreas; Beck, Michael; Roscher, Adelbert A.; Muntau, Ania C. (2007). „Disease manifestations and X inactivation in heterozygous females with Fabry disease”. Acta Paediatrica. 95 (451): 30—38. PMID 16720462. S2CID 15437239. doi:10.1111/j.1651-2227.2006.tb02386.x..
- ^ a b v Mehta, A.; Beck, M.; Sunder-Plassmann, G.; Gal, A.; Schäfer, E.; Rohard, I. (2006). „The genetic basis of Fabry disease”. PMID 21290673.
- ^ A. Gal: Molecular genetics of Fabry disease and Genotype-phenotype correlation. In: D. Elstein, G. Altarescu, M. Beck (eds.): Fabry disease. Verlag Springer. Elstein, Deborah; Altarescu, Gheona; Beck, Michael (2010). Fabry Disease. Springer. str. 3—19. ISBN 978-90-481-9032-4..
- ^ R. O. Brady: Fabry Disease – An Overview. In: D. Elstein, G. Altarescu, M. Beck (eds.): Fabry Disease. Verlag Springer. Elstein, Deborah; Altarescu, Gheona; Beck, Michael (2010). Fabry Disease. Springer. ISBN 978-90-481-9032-4.
- ^ Futerman, Anthony H.; Van Meer, Gerrit (2004). „The cell biology of lysosomal storage disorders”. Nature Reviews Molecular Cell Biology. 5 (7): 554—565. PMID 15232573. S2CID 21605400. doi:10.1038/nrm1423. hdl:1874/14471.
- ^ Vedder, Anouk C.; Linthorst, Gabor E.; Houge, Gunnar; Groener, Johannna E.M.; Ormel, Els E.; Bouma, Berto J.; Aerts, Johannes M.F.G.; Hirth, Asle; Hollak, Carla E.M. (2007). „Treatment of Fabry Disease: Outcome of a Comparative Trial with Agalsidase Alfa or Beta at a Dose of 0.2 mg/Kg”. PLOS ONE. 2 (7): e598. Bibcode:2007PLoSO...2..598V. PMC 1913555 . PMID 17622343. doi:10.1371/journal.pone.0000598 .
- ^ a b Vedder, A. C.; Linthorst, G. E.; Van Breemen, M. J.; Groener, J. E. M.; Bemelman, F. J.; Strijland, A.; Mannens, M. M. A. M.; Aerts, J. M. F. G.; Hollak, C. E. M. (2007). „The Dutch Fabry cohort: Diversity of clinical manifestations and Gb3 levels”. Journal of Inherited Metabolic Disease. 30 (1): 68—78. PMID 17206462. S2CID 10333874. doi:10.1007/s10545-006-0484-8..
- ^ Sanchez-Nino, M. D.; Sanz, A. B.; Carrasco, S.; Saleem, M. A.; Mathieson, P. W.; Valdivielso, J. M.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. (2011). „Globotriaosylsphingosine actions on human glomerular podocytes: Implications for Fabry nephropathy”. Nephrology Dialysis Transplantation. 26 (6): 1797—1802. PMID 20504837. doi:10.1093/ndt/gfq306..
- ^ A. P. Burlina, K. B. Sims u. a.: Early diagnosis of peripheral nervous system involvement in Fabry disease and treatment of neuropathic pain: the report of an expert panel. In: BMC neurology. Band 11, 2011, S. 61
- ^ „Angiokeratom • LekarInfo” (na jeziku: srpski). 2014-08-26. Pristupljeno 2023-02-07.
- ^ R Trickett and H Dowd Angiokeratoma of the scrotum: a case of scrotal bleeding Emerg Med J. 2006 Oct; 23(10): e57.
- ^ a b Shah, Jaymin S.; Hughes, Derralynn A.; Sachdev, Bhavesh; Tome, Maite; Ward, Deirdre; Lee, Philip; Mehta, Atul B.; Elliott, Perry M. (2005). „Prevalence and Clinical Significance of Cardiac Arrhythmia in Anderson-Fabry Disease”. The American Journal of Cardiology. 96 (6): 842—846. PMID 16169374. doi:10.1016/j.amjcard.2005.05.033..
- ^ Senechal, M.; Germain, D. P. (2003). „Fabry disease: A functional and anatomical study of cardiac manifestations in 20 hemizygous male patients”. Clinical Genetics. 63 (1): 46—52. PMID 12519371. S2CID 11602510. doi:10.1034/j.1399-0004.2003.630107.x.
- ^ Elliott, P. M.; Kindler, H.; Shah, J. S.; Sachdev, B.; Rimoldi, O. E.; Thaman, R.; Tome, M. T.; McKenna, W. J.; Lee, P.; Camici, P. G. (2005). „Coronary microvascular dysfunction in male patients with Anderson-Fabry disease and the effect of treatment with galactosidase A”. Heart. 92 (3): 357—360. PMC 1860797 . PMID 16085718. doi:10.1136/hrt.2004.054015.
- ^ Moon, J. C.; Sheppard, M.; Reed, E.; Lee, P.; Elliott, P. M.; Pennell, D. J. (2006). „The histological basis of late gadolinium enhancement cardiovascular magnetic resonance in a patient with Anderson-Fabry disease”. Journal of Cardiovascular Magnetic Resonance : Official Journal of the Society for Cardiovascular Magnetic Resonance. 8 (3): 479—482. PMID 16755835. doi:10.1080/10976640600605002.
- ^ Linhart, A.; Kampmann, C.; Zamorano, J. L.; Sunder-Plassmann, G.; Beck, M.; Mehta, A.; Elliott, P. M.; European FOS Investigators (2007). „Cardiac manifestations of Anderson-Fabry disease: Results from the international Fabry outcome survey”. European Heart Journal. 28 (10): 1228—1235. PMID 17483538. doi:10.1093/eurheartj/ehm153..
- ^ Takenaka, Toshihiro; Teraguchi, Hiroyuki; Yoshida, Aichi; Taguchi, Syuhei; Ninomiya, Kenjiro; Umekita, Yoshihisa; Yoshida, Hiroki; Horinouchi, Michiko; Tabata, Kazuhiro; Yonezawa, Suguru; Yoshimitsu, Makoto; Higuchi, Koji; Nakao, Shoichiro; Anan, Ryuichiro; Minagoe, Shinichi; Tei, Chuwa (2008). „Terminal stage cardiac findings in patients with cardiac Fabry disease: An electrocardiographic, echocardiographic, and autopsy study”. Journal of Cardiology. 51 (1): 50—59. PMID 18522775. doi:10.1016/j.jjcc.2007.12.001..
- ^ Linhart, Aleš; Paleček, Tomáš; Bultas, Jan; Ferguson, James J.; Hrudová, Jana; Karetová, Debora; Zeman, Jiři; Ledvinová, Jana; Poupětová, Helena; Elleder, Milan; Aschermann, Michael (2000). „New insights in cardiac structural changes in patients with Fabry's disease”. American Heart Journal. 139 (6): 1101—1108. PMID 10827394. doi:10.1067/mhj.2000.105105..
- ^ Pieroni, Maurizio; Chimenti, Cristina; Ricci, Roberta; Sale, Patrizio; Russo, Matteo Antonio; Frustaci, Andrea (2003). „Early Detection of Fabry Cardiomyopathy by Tissue Doppler Imaging”. Circulation. 107 (15): 1978—1984. PMID 12668521. S2CID 2902358. doi:10.1161/01.CIR.0000061952.27445.A0..
- ^ Pieroni, M.; Chimenti, C.; Russo, A.; Russo, M. A.; Maseri, A.; Frustaci, A. (2004). „Tissue Doppler imaging in Fabry disease”. Current Opinion in Cardiology. 19 (5): 452—457. PMID 15316452. S2CID 20093987. doi:10.1097/01.hco.0000131534.25034.43.
- ^ Niemann, M.; Breunig, F.; Beer, M.; Herrmann, S.; Strotmann, J.; Hu, K.; Emmert, A.; Voelker, W.; Ertl, G.; Wanner, C.; Weidemann, F. (2010). „The right ventricle in Fabry disease: Natural history and impact of enzyme replacement therapy”. Heart. 96 (23): 1915—1919. PMID 20965976. S2CID 2837188. doi:10.1136/hrt.2010.204586..
- ^ Kampmann, C.; Baehner, F. A.; Whybra, C.; Bajbouj, M.; Baron, K.; Knuf, M.; Wiethoff, C. M.; Trübel, H.; Beck, M. (2005). „The right ventricle in Fabry disease”. Acta Paediatrica (Oslo, Norway : 1992). Supplement. 94 (447): 15—8; discussion 9—10. PMID 15895706. S2CID 24554620. doi:10.1111/j.1651-2227.2005.tb02104.x.
- ^ a b Sims, Katherine; Politei, Juan; Banikazemi, Maryam; Lee, Philip (2009). „Stroke in Fabry Disease Frequently Occurs Before Diagnosis and in the Absence of Other Clinical Events”. Stroke. 40 (3): 788—794. PMID 19150871. S2CID 9265154. doi:10.1161/STROKEAHA.108.526293..
- ^ Fellgiebel, Andreas; Müller, Matthias J.; Ginsberg, Lionel (2006). „CNS manifestations of Fabry's disease”. The Lancet Neurology. 5 (9): 791—795. PMID 16914407. S2CID 38935531. doi:10.1016/S1474-4422(06)70548-8..
- ^ Magage, S.; Lubanda, J.‐C.; Susa, Z.; Bultas, J.; Karetová, D.; Dobrovolný, R.; Hřebíček, M.; Germain, D. P.; Linhart, A. (2007). „Natural history of the respiratory involvement in Anderson–Fabry disease”. Journal of Inherited Metabolic Disease. 30 (5): 790—799. PMID 17619837. S2CID 11621381. doi:10.1007/s10545-007-0616-9..
- ^ Degraba, T.; Azhar, S.; Dignat-George, F.; Brown, E.; Boutière, B.; Altarescu, G.; McCarron, R.; Schiffmann, R. (2000). „Profile of endothelial and leukocyte activation in Fabry patients”. Annals of Neurology. 47 (2): 229—233. PMID 10665494. S2CID 19202089. doi:10.1002/1531-8249(200002)47:2<229::AID-ANA13>3.0.CO;2-T.
- ^ Palla, A.; Hegemann, S.; Widmer, U.; Straumann, D. (2007). „Vestibular and auditory deficits in Fabry disease and their response to enzyme replacement therapy” (PDF). Journal of Neurology. 254 (10): 1433—1442. PMID 17934877. S2CID 139765. doi:10.1007/s00415-007-0575-y..
- ^ Sakurai, Yuika; Kojima, Hiromi; Shiwa, Masanori; Ohashi, Toya; Eto, Yoshikatsu; Moriyama, Hiroshi (2009). „The hearing status in 12 female and 15 male Japanese Fabry patients”. Auris Nasus Larynx. 36 (6): 627—632. PMID 19261412. doi:10.1016/j.anl.2009.01.001..
- ^ Germain, D. P.; Avan, P.; Chassaing, A.; Bonfils, P. (2002). „Patients affected with Fabry disease have an increased incidence of progressive hearing loss and sudden deafness: An investigation of twenty-two hemizygous male patients”. BMC Medical Genetics. 3: 10. PMC 134464 . PMID 12377100. doi:10.1186/1471-2350-3-10 .
- ^ Rosenberg, D. M.; Ferrans, V. J.; Fulmer, J. D.; Line, B. R.; Barranger, J. A.; Brady, R. O.; Crystal, R. G. (1980). „Chronic airflow obstruction in Fabry's disease”. The American Journal of Medicine. 68 (6): 898—905. PMID 6247911. doi:10.1016/0002-9343(80)90224-7.
- ^ Germain, D. P.; Benistan, K.; Boutouyrie, P.; Mutschler, C. (2005). „Osteopenia and osteoporosis: Previously unrecognized manifestations of Fabry disease”. Clinical Genetics. 68 (1): 93—95. PMID 15952993. S2CID 40653911. doi:10.1111/j.1399-0004.2005.00457.x..
- ^ Mersebach, Henriette; Johansson, Jan-Ove; Rasmussen, åse Krogh; Bengtsson, Bengt-Åke; Rosenberg, Kirsten; Hasholt, Lis; Sørensen, Sven Asger; Sørensen, Søren Schwartz; Feldt-Rasmussen, Ulla (2007). „Osteopenia: A common aspect of Fabry disease. Predictors of bone mineral density”. Genetics in Medicine. 9 (12): 812—818. PMID 18091430. S2CID 22998638. doi:10.1097/GIM.0b013e31815cb197..
- ^ R. O. Brady: Bone and muscle involvement in Fabry disease. In: D. Elstein, G. Altarescu, M. Beck (eds.): Fabry Disease. Verlag Springer. Elstein, Deborah; Altarescu, Gheona; Beck, Michael (2010). Fabry Disease. Springer. str. 293—298. ISBN 978-90-481-9032-4.eingeschränkte Vorschau in der Google-Buchsuche
- ^ Grau, A. J.; Schwaninger, M.; Goebel, H. H.; Beck, M. (2003). „Morbus Fabry”. Der Nervenarzt. 74 (6): 489—496. PMID 12799787. S2CID 41809661. doi:10.1007/s00115-003-1513-6..
- ^ Marchesoni, Cintia L.; Roa, Norma; Pardal, Ana María; Neumann, Pablo; Cáceres, Guillermo; Martínez, Pablo; Kisinovsky, Isaac; Bianchi, Silvia; Tarabuso, Ana Lía; Reisin, Ricardo C. (2010). „Misdiagnosis in Fabry Disease”. The Journal of Pediatrics. 156 (5): 828—831. PMID 20385321. doi:10.1016/j.jpeds.2010.02.012..
- ^ Mehta, A.; Ricci, R.; Widmer, U.; Dehout, F.; Garcia De Lorenzo, A.; Kampmann, C.; Linhart, A.; Sunder-Plassmann, G.; Ries, M.; Beck, M. (2004). „Fabry disease defined: Baseline clinical manifestations of 366 patients in the Fabry Outcome Survey”. European Journal of Clinical Investigation. 34 (3): 236—242. PMID 15025684. S2CID 40805388. doi:10.1111/j.1365-2362.2004.01309.x..
- ^ a b MacDermot, K. D.; Holmes, A.; Miners, A. H. (2001). „Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 98 hemizygous males”. Journal of Medical Genetics. 38 (11): 750—760. PMC 173476 . PMID 11694547. doi:10.1136/jmg.38.11.750.
- ^ „Morbus Fabry ist selten, die Symptomatik unspezifisch - Vor der Diagnose steht oft eine Odyssee von”. AerzteZeitung.de (na jeziku: nemački). 2004-12-15. Pristupljeno 2022-02-23.
- ^ Mayes, J. S.; Scheerer, J. B.; Sifers, R. N.; Donaldson, M. L. (1981). „Differential assay for lysosomal alpha-galactosidases in human tissues and its application to Fabry's disease”. Clinica Chimica Acta; International Journal of Clinical Chemistry. 112 (2): 247—251. PMID 6263521. doi:10.1016/0009-8981(81)90384-3.
- ^ Hoffmann, Bjoern; Georg Koch, Hans; Schweitzer-Krantz, Susanne; Wendel, Udo; Mayatepek, Ertan (2005). „Deficient α-galactosidase a activity in plasma but no Fabry disease – a pitfall in diagnosis”. Clinical Chemistry and Laboratory Medicine (CCLM). 43 (11): 1276—1277. PMID 16232095. S2CID 85843657. doi:10.1515/CCLM.2005.219..
- ^ Linthorst, Gabor E.; Vedder, Anouk C.; Aerts, Johannes M.F.G.; Hollak, Carla E.M. (2005). „Screening for Fabry disease using whole blood spots fails to identify one-third of female carriers”. Clinica Chimica Acta. 353 (1): 201—203. PMID 15698608. doi:10.1016/j.cccn.2004.10.019..
- ^ a b B. E. Smid, L. van der Tol, M. Biegstraaten, G. E. Linthorst, C. E. M. Hollak, B. J. H. M. Poorthuis: Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease. In: J Med Genet. Band 52, Nr. 4, 2015, S. 262–226.
- ^ Roy, S.; Gaudin, K.; Germain, D.P.; Baillet, A.; Prognon, P.; Chaminade, P. (2004). „Optimisation of the separation of four major neutral glycosphingolipids: Application to a rapid and simple detection of urinary globotriaosylceramide in Fabry disease”. Journal of Chromatography B. 805 (2): 331—337. PMID 15135109. doi:10.1016/j.jchromb.2004.03.037.
- ^ Desnick, Robert J. (2007). „Prenatal diagnosis of Fabry disease”. Prenatal Diagnosis. 27 (8): 693—694. PMID 17533632. S2CID 33020619. doi:10.1002/pd.1767..
- ^ A. M. Das: Angeborene Stoffwechselstörungen. In: A. Rieder, B. Lohff (eds.): Gender Medizin. Verlag Springer. Rieder, Anita; Lohff, Brigitte (2004). Gender Medizin: Geschlechtsspezifische Aspekte für die klinische Praxis. Springer. str. 67f. ISBN 3-211-00766-0..
- ^ a b Kasper, David C.; Herkner, Kurt R.; Streubel, Berthold; Item, Chike B.; Ratschmann, Rene; Herman, Joseph L.; Shushan, Bori; Martin, Monica; Orsini, Joseph J.; Mechtler, Thomas P.; Metz, Thomas F. (2011). „Simplified Newborn Screening Protocol for Lysosomal Storage Disorders”. Clinical Chemistry. 57 (9): 1286—1294. PMID 21771947. doi:10.1373/clinchem.2011.164640..
- ^ M. Spada, D. Kasper, V. Pagliardini, E. Biamino, S. Giachero, F. Porta: Metabolic progression to clinical phenotype in classic Fabry disease. In: Ital J Pediatr. Band 43, Nr. 1, 2017, S. 1.
- ^ Pastores, G. M. (2006). „Miglustat: Substrate reduction therapy for lysosomal storage disorders associated with primary central nervous system involvement”. Recent Patents on CNS Drug Discovery. 1 (1): 77—82. PMID 18221193. doi:10.2174/157488906775245282.
- ^ „Therapie des Morbus Fabry”. 2015-02-03. Arhivirano iz originala 03. 02. 2015. g. Pristupljeno 2022-02-24.
- ^ a b Thomaidis, Thomas; Relle, Manfred; Reinke, Joerg; Beck, Michael; Schwarting, Andreas (2009). „Wirkung der Enzymersatztherapie (ERT) auf die Nierenfunktion von Patienten mit Morbus Fabry”. Medizinische Klinik. 104 (9): 699—703. PMID 19779674. S2CID 7132747. doi:10.1007/s00063-009-1152-1..
- ^ „March on, not in”. Nature Medicine. 17 (5): 515. 2011. PMID 21546944. S2CID 205378860. doi:10.1038/nm0511-515..
- ^ Beck, Michael (2002). „Agalsidase alfa – a preparation for enzyme replacement therapy in Anderson–Fabry disease”. Expert Opinion on Investigational Drugs. 11 (6): 851—858. PMID 12036428. S2CID 37196144. doi:10.1517/13543784.11.6.851.
- ^ Lee, K.; Jin, X.; Zhang, K.; Copertino, L.; Andrews, L.; Baker-Malcolm, J.; Geagan, L.; Qiu, H.; Seiger, K.; Barngrover, D.; McPherson, J. M.; Edmunds, T. (2003). „A biochemical and pharmacological comparison of enzyme replacement therapies for the glycolipid storage disorder Fabry disease”. Glycobiology. 13 (4): 305—313. PMID 12626384. doi:10.1093/glycob/cwg034..
- ^ Keating, G. M.; Simpson, D. (2007). „Spotlight on agalsidase beta in Fabry disease”. BioDrugs. 21 (4): 269—271. PMID 17628124. S2CID 26215578. doi:10.2165/00063030-200721040-00007.
- ^ Vedder, Anouk C.; Linthorst, Gabor E.; Houge, Gunnar; Groener, Johannna E.M.; Ormel, Els E.; Bouma, Berto J.; Aerts, Johannes M.F.G.; Hirth, Asle; Hollak, Carla E.M. (2007). „Treatment of Fabry Disease: Outcome of a Comparative Trial with Agalsidase Alfa or Beta at a Dose of 0.2 mg/Kg”. PLOS ONE. 2 (7): e598. Bibcode:2007PLoSO...2..598V. PMC 1913555 . PMID 17622343. doi:10.1371/journal.pone.0000598 .
- ^ Ohashi, Toya; Sakuma, Mio; Kitagawa, Teruo; Suzuki, Ken; Ishige, Nobuyuki; Eto, Yoshikatsu (2007). „Influence of antibody formation on reduction of globotriaosylceramide (GL-3) in urine from Fabry patients during agalsidase beta therapy”. Molecular Genetics and Metabolism. 92 (3): 271—273. PMID 17689998. doi:10.1016/j.ymgme.2007.06.013..
- ^ Arends, Maarten; Biegstraaten, Marieke; Wanner, Christoph; Sirrs, Sandra; Mehta, Atul; Elliott, Perry M.; Oder, Daniel; Watkinson, Oliver T.; Bichet, Daniel G.; Khan, Aneal; Iwanochko, Mark; Vaz, Frédéric M.; Van Kuilenburg, André B P.; West, Michael L.; Hughes, Derralynn A.; Hollak, Carla E M. (2018). „Agalsidase alfa versus agalsidase beta for the treatment of Fabry disease: An international cohort study”. Journal of Medical Genetics. 55 (5): 351—358. PMC 5931248 . PMID 29437868. S2CID 19054248. doi:10.1136/jmedgenet-2017-104863..
- ^ U. Fricke, U. Schwabe: Neue Arzneimittel 2007. In: U. Schwabe, D. Paffrath (eds.): Arzneiverordnungs-Report 2008. Verlag Springer, 2008, S. 74. eingeschränkte Vorschau in der Google-Buchsuche
- ^ Hoffmann, Björn; Mayatepek, Ertan (2009). „Fabry Disease”. Deutsches Ärzteblatt International. 106 (26): 440—447. PMC 270439 . PMID 19623315. doi:10.3238/arztebl.2009.0440.
- ^ Watt, Torquil; Burlina, Alessandro P.; Cazzorla, Chiara; Schönfeld, Dorothee; Banikazemi, Maryam; Hopkin, Robert J.; Martins, Ana Maria; Sims, Katherine; Beitner-Johnson, Dana; O'Brien, Fanny; Feldt-Rasmussen, Ulla (2010). „Agalsidase beta treatment is associated with improved quality of life in patients with Fabry disease: Findings from the Fabry Registry”. Genetics in Medicine. 12 (11): 703—712. PMID 20885332. S2CID 23666715. doi:10.1097/GIM.0b013e3181f13a4a..
- ^ Filling-Katz, M. R.; Merrick, H. F.; Fink, J. K.; Miles, R. B.; Sokol, J.; Barton, N. W. (1989). „Carbamazepine in Fabry's disease: Effective analgesia with dose-dependent exacerbation of autonomic dysfunction”. Neurology. 39 (4): 598—600. PMID 2494569. S2CID 38971405. doi:10.1212/wnl.39.4.598.
- ^ National Institute for Health and Clinical Excellence: Neuropathic pain: The pharmacological management of neuropathic pain in adults in non-specialist settings (NICE clinical guideline 96). Centre for Clinical Practice at NICE; 2010.
- ^ Cruccu, G.; Sommer, C.; Anand, P.; Attal, N.; Baron, R.; Garcia-Larrea, L.; Haanpaa, M.; Jensen, T. S.; Serra, J.; Treede, R. -D. (2010). „EFNS guidelines on neuropathic pain assessment: Revised 2009”. European Journal of Neurology. 17 (8): 1010—1018. PMID 20298428. S2CID 2393393. doi:10.1111/j.1468-1331.2010.02969.x..
- ^ Argoff, C. E.; Barton, N. W.; Brady, R. O.; Ziessman, H. A. (1998). „Gastrointestinal symptoms and delayed gastric emptying in Fabry's disease: Response to metoclopramide”. Nuclear Medicine Communications. 19 (9): 887—891. PMID 10581595. doi:10.1097/00006231-199809000-00009.
- ^ Tahir, Hindia; Jackson, Leslie L.; Warnock, David G. (2007). „Antiproteinuric Therapy and Fabry Nephropathy”. Journal of the American Society of Nephrology. 18 (9): 2609—2617. PMID 17656478. doi:10.1681/ASN.2006121400..
- ^ Stetter, Christian E. (2007). In vivo Untersuchung des kardialen Stoffwechsels bei Morbus Fabry mittels 31Phosphor-MR-Spektroskopie (Teza). Universität Würzburg.
Literatura uredi
- Tucić, N, Matić, Gordana: O genima i ljudima, Centar za primenjenu psihologiju, Beograd, 2002.
- Marinković, D, Tucić, N, Kekić, V: Genetika, Naučna knjiga, Beograd
- Tatić, S, Kostić, G, Tatić, B: Humani genom, ZUNS, Beograd, 2002.
- Matić, Gordana: Osnovi molekularne biologije, Zavet, Beograd, 1997.
- Ridli, M: Genom - autobiografija vrste u 23 poglavlja, Plato, Beograd, 2001.
- Prentis S: Biotehnologija, Školska knjiga, Zagreb, 1991.
- Dumanović, J, marinković, D, Denić, M: Genetički rečnik, Beograd, 1985.
- Kosanović, M, Diklić, V: Odabrana poglavlja iz humane genetike, Beograd, 1986.
- Švob, T. i sradnici: Osnovi opće i humane genetike, Školska knjiga, Zagreb, 1990.
Spoljašnje veze uredi
| Klasifikacija | |
|---|---|
| Spoljašnji resursi |
- „Fabry Disease Information Page”. Arhivirano iz originala 02. 12. 2016. g. at NINDS (jezik: engleski)
- Fabry disease at NLM Genetics Home Reference (jezik: engleski)
 | Molimo Vas, obratite pažnju na važno upozorenje u vezi sa temama iz oblasti medicine (zdravlja). |