Фабријева болест
Фабријева болест (акроним ФБ) или Андерсон–Фабријева болест представља за X везано, генетски условљено обољење лизозима код које долази до накупљања гликосфинголипида на унутрашњем слоју крвних судова (ендотелу), на бројним унутрашњим органима, на њиховим епителима и глатко-мишићним структурама.[1]
| Фабријева болест | |
|---|---|
| Синоними | Fabry's disease, Anderson–Fabry disease, angiokeratoma corporis diffusum, alpha-galactosidase A deficiency |
 | |
| Алфа галактозид - дефицитаран протеина код Фабријеве болести | |
| Изговор | |
| Специјалности | ендокринологија, кардиологија, нефрологија, дерматологија |
| Компликације | оштећење срца, срчане аритмије |
| Време појаве | детињство |
| Узроци | генетика |
| Дијагностички метод | тест активности ензима, генетско тестирање |
| Лечење | замена ензима |
Фабријева болест је типичан представник ретких болести, која за разлику од многих других тренутно има много повољнију прогнозу, након увођења супституционе ензимске терапије и развоја бољих клиничких, биохемијских и молекуларних дијагностичким метода које могу значајно да скрате време препознавање ових пацијената.[2]
У зависности од органа који су захваћени, могу се појавити веома различити симптоми. Појединачно веома различита манифестација болести и њена реткост знатно отежавају дијагнозу , обично се исправно поставља тек много година након појаве првих симптома.
Болест првенствено погађа мушкарце, али хетерозиготне (мешане крви) жене такође могу да оболе. Код њих је, међутим, болест обично мање тешка и тек у средњим годинама почиње да постаје клинички релевантна. Квалитет живота пацијената који пате од Фабријеве болести је често доста оштећен.
Болест се узрочно лечи терапијом замене ензима од 2001. Пацијенти добијају генетски модификовану α-галактозидазу А као инфузију до краја живота . Тренутно не постоји лек за Фабријеву болест. Ако се не лече, мушки пацијенти живе до просечне старости од око 50, а жене до око 70 година. Главни узроци ране смртности су хронична бубрежна инсуфицијенција , оштећење срца и оштећено доток крви у мозак .
Активност ензима α-галактозидазе А је у великој мери смањена мутацијом ( генетском модификацијом) на Х хромозому (полни хромозом) , одговоран је за разградњу масти. У лизозомима (центри за рециклажу ћелија), метаболички производ глоботриаозилцерамид (који се такође назива Гб3 или ГЛ-3), гликосфинголипид , више не може да се разгради у довољној мери. Гб3 се акумулира првенствено у ћелијама које облажу унутрашњост крвних судова , ендотелним ћелијама, па како болест напредује, ове акумулације постају патолошке , односно изазивају Фабријеву болест. У зависности од тока болести, то понекад може потрајати деценијама.
Историја
уредиКао посебан клинички ентитет, Фабријеву болест је први пут описао енглески хирург Вилијам Андерсон 1897. године.[1]
Годину дана касније (1898. године), немачки дерматолог Јохан Фабри је описао дифузне ангиокератоме у 13 година старог дечака са протеинуријом, који су такође боловали од ове болест, па је је овај епоним по имену Јоханесa Фабријa назван Фабријева болест,[1] мада се у литератури ова болст назива и Андерсон-Фабријева болест.[3][4]
Епидемиологија
уредиУчесталос Фабријева болести се креће од. 1 50 000(1:40 000 до. 1 117 000) иако су блажи облици болести који се јављају касније у животу јављају нешто чешће.[5][6][7] Међутим, новије студије засноване на подацима са скрининга новорођенчади указују на значајно већу учесталост Фабријеве болести. Од 2004. до 2006. године, преваленција од око 1:3.100 на Тајвану[8], а око 1:1.500 код мушке новорођенчади у северној Италији, или са преваленцом од 1:3.100, која резултује са 26.450 пацијената са Фабријевом болешћу у Немачкој.[9]
Претпоставља се да код многих пацијената болест остаје непрепозната током њиховог живота и да се превремена смрт приписује другим болестима, као што су изоловане кардиомиопатије попут оних које се налазе код пацијената са Фабријевом болешћу са резидуалном активношћу α-галактозидазе А.[8] Још један показатељ овога је да је на Тајвану 86% новорођенчади која су била позитивна имала криптичну мутацију спајањатипа ИВС4+919Г > А, који је раније пронађен првенствено код пацијената са Фабријевом болешћу са срчаним фенотипом.[10] Код ових пацијената, Фабријева болест се у суштини манифестује болешћу миокарда (кардиомиопатија). Интронички облик ове мутације присутан је код многих тајванских пацијената са хипертрофичном кардиомиопатијом.[11][12]
У одређеним субпопулацијама повезаним са симптомима Фабријеве болести, инциденција је сама по себи већа. Студија на 911 шпанских пацијената на хемодијализи идентификовала је четири мушкарца и три жене са абнормалностима у ГЛА гену, што одговара преваленцији од. 1 182у овој субпопулацији.[13]
Етиологија
уредиФабријева болест је за Х везана наследна лизозомска болест депоновања заснована на лизозомском недостатку ензим алфа галактозидаза (АГАЛ-А) одговорног за хидролизу галактозе остатка сфинголипида због чега су они (попут глоботриазилцерамида, галабиозил церамида и глоботриазилсфингозин, сфингозин 1 фосфат) акумулирају у бројним ћелије и ткива.[1] Ова акумулација остатка сфинголипида резултује прогресивним оштећењем ткива и органским оштећења која су најчешће последица промена у:[3][1]
- ендотелу и глатких мишићних ћелија микроциркулације,
- акумулацији метаболита сфинголпида у ћелијама срца, бубрега, рожњаче, панкреаса, црева, плућа, панкреаса и
- разних врста нервних ћелија централног и периферног нервног система.[14]
Генетика и молекуларна биологија
уреди

А Седамнаестогодишња девојка са трансверзијом тимина у гванин у ексону 6, позиција 884. Ова супституција нуклеотида мења кодон ТТЦ, који кодира аминокиселину фенилаланин , у ТГЦ, што се преводи у инкорпорацију цистеина у производ гена α-галактозидазе.
А. Сходно томе, у α-галактозидази А овог пацијента, налази се цистеин на позицији 295 уместо фенилаланина. Постоји мутација п.Пхе295Цис.
Б. 46-годишња жена са Т-то-Г трансверзијом у свом ГЛА гену на егзону 1, позиција 125. АТГ кодон, који кодира метионин , стога постаје АГГ, што представља аминокиселину аргинин , која се затим налази на позицији 42 α-галактозидазе А након транслације. То је п.Мет42Арг мутација.
Ц Овај 63-годишњи пацијент има ГТ трансверзију у ексону 6, позиција 982. Као резултат размене нуклеотида, ГГГ кодон постаје ТГГ, а глицин постаје триптофан , који се затим налази на позицији 328 α-галактозидазе А. Мутација је стога означена као п.Гли328Трп.

Фабријева болест је болест заснована на генетском дефекту (мутацији) у женском полном хромозому , Х-хромозому. Сваки отац погођен болешћу преноси болест на све своје ћерке, док сви његови синови остају здрави. Ако мајка носи мутирани ген, њена деца – без обзира на пол – имају 50% ризик да наследе болест. Мутација утиче на ГЛА ген, који се налази на дугом краку X хромозома на локусу гена к22.1.[15]
Генски производ је хомодимер и, као и сви лизозомални ензими, обезбеђен је са остатком маноза-6-фосфата котранслационо, односно током транслације мРНК у секвенцу аминокиселина . Неке од фосфорилисаних молекула α-галактозидазе А луче ћелије и преузимају их друге ћелије преко рецептора маноза-6-фосфата везаног за мембрану ендоцитозома. Поновно преузимање фосфорилисане α-галактозидазе А преко маноза-6-фосфатног рецептора је основа за терапију замене ензима.[16]
Посебности наслеђа везаног за Х
уредиЗбог Х-хромозомског наслеђа, болест се различито манифестује код мушкараца и жена. Мушки пацијенти се називају хемизиготи , а жене хетерозиготни носиоци. Претпостављало се да само мушкарци могу да развију Фабријеву болест и да су хетерозиготне жене само носиоци. Ово је случај са великом већином других наследних болести повезаних са Х- као што су хемофилија или Дишенова мишићна дистрофија. Сада је познато да жене које су хетерозиготе за ову карактеристику такође могу развити Фабријеву болест. Неки аутори стога препоручују избегавање термина Х-везано рецесивно , јер је то погрешно. Уместо тога, препоручује се терминологија Х-везано наслеђивање.[17][18][19]
Код хетерозиготних пацијената, један немутирани и један мутирани Х хромозом присутни су у свакој ћелији тела са ДНК са језгром. Инактивација Х инактивира један од два Х хромозома у свакој ћелији. Ова инактивација се дешава независно у свакој ћелији и по принципу случајности (мозаик ). Према томе, статистички гледано, 50% ћелија ће производити α-галактозидазу А са мало или без активности, у зависности од врсте мутације. Осталих 50% ћелија производи α-галактозидазу А са нормалном активношћу („здраве ћелије“). Део активне α-галактозидазе А производе „мутантне ћелије“ са активираним Х хромозомом, на којима је место дефектног ГЛА гена, као што је претходно описано, заузето ендоцитозом. Иако је овај пренос ензима довољан да спречи имуни систем да елиминише мутиране ћелије , он је пренизак да би се компензовао дефект гена како би се спречило накупљање глоботриаозилцерамида.[20] У поређењу са другим лизозомалним ензимима, унос α-галактозидазе А преносом ензима је релативно низак.[21]
Са Х-инактивацијом се може објаснити да у просеку код хетерозиготних жена болест постаје симптоматска много касније и мање је изражена него код мушкараца. Међутим, то није довољан модел за разумевање широког спектра различитих манифестација болести код жена. На пример, око 10% пацијената захтева терапију замене бубрега током прогресије болести, што одговара „класичном фенотипу“ код мушкараца. Друге хетерозиготне жене, с друге стране, остају углавном без симптома. Разлог за то још није у потпуности разјашњен.[11]
Једна хипотеза је да неправилности у инактивацији Х хромозома играју важну улогу у опсегу варијација хетерозиготног фенотипа. Ово се назива „искривљена“ Х инактивација, у којој је статистички очекивани однос. 50 50између „мутантних“ и „здравих“ ћелија јасно померен. Овај помак није узрокован предношћу раста мутантних ћелија. Индикација искривљене Х инактивације су хетерозиготни пацијенти са фенотипом код којих је Фабријева болест у потпуности развијена, пошто је мутирани Х хромозом активиран у преко 95% ћелија. Отприлике један од 200 пацијената са Фабријевом болешћу има овај фенотип. Још један показатељ искривљене Х-инактивације је женски хетерозиготни пар монозиготних близанаца , где је један од близанаца асимптоматски, а други је клинички релевантан.[22]
У студији са 28 пацијената, код већине испитаника је пронађена искривљена инактивација Х у леукоцитима откривено, али није било повезано са клиничком манифестацијом болести или са заосталом активношћу ензима. Аутори стога не виде никакву везу између фенотипа и искривљене Х активације.[23]
Мутационе варијанте
уредиДефекти ГЛА гена узроковани мутацијама су веома хетерогени. До сада је забележено преко 500 различитих мутација. То укључује смислене и бесмислене тачкасте мутације, мутације спајања, мале делеције и уметања, и велике делеције.[11] Тачкасте мутације су најчешће (око 71%), праћене малим делецијама и инсерцијама које утичу на мање од 60 нуклеотида (око 27%) и великим делецијама које утичу на један или више егзона (око 2%).[24]
У већини случајева, мутација доводи до потпуног губитка активности ензима.[25]
Неке мутације које доводе до промена α-галактозидазе А и које су довољно удаљене од активног места ензима доводе само до малих структурних промена у ензиму, тако да је одређена резидуална активност ензима и даље присутна. Такве мутације као што су п.Мет72Вал, п.Глн279Глу или п.Мет296Иле карактерише благи фенотип болести.[24]
На ГЛА гену нема изражене жаришне тачке, то је подручје посебно подложно мутацијама. Упадљиве су честе реаранжмане ДНК у егзону 7, који очигледно има повећану подложност преуређивању.[24]
Патологија
уредиФабријева болест спада у групу лизозомалних болести складиштења, која обухвата најмање 50 чланова, и ту у подгрупу сфинголипидоза . Болест је заснована на недостатку лизозомалног ензима α-галактозидазе А. Због овог недостатка, одређени метаболички производи као што је глоботриаозилцерамид (Гб3, Гл3, раније називан и церамид трихексозид) акумулирају се у ендотелним ћелијама различитих система органа. Смањена активност α-галактозидазе у суштини доводи до акумулације глоботриаозилцерамида. Поред тога, акумулирају се дигалактозаилцерамид (посебно у бубрезима) и глоботриаозилшингозин (лизо-Гб3, лизо-Гл3).[26] Ови сфинголипиди су важне компоненте ћелијске мембране .
Тачне везе између смањене или чак потпуно нестале активности α-галактозидазе А и патолошких процеса у захваћеним органима – који на крају доводе до Фабријеве болести – још увек нису довољно разјашњене. Разноврсност захваћених органа сугерише да секундарни биохемијски механизми који укључују сфинголипиде одређују ток болести.[27]
Симптоми описани у следећем поглављу, као што је прогресивна хронична бубрежна инсуфицијенција , у многим публикацијама се приписују акумулацији глоботриаозилцерамида у лизозому ендотелних ћелија. Међутим, бројни клинички ефекти, посебно у терапији замене ензима за Фабријеву болест, не одговарају овом очигледно поједностављеном моделу. На пример, код неких пацијената се могу приметити прогресивне компликације , што сугерише да не постоји директна корелација између Гб3 и клиничких манифестација Фабријеве болести.[28] Запажање да је велики проценат жена ГЛА- Носиоци мутације развијају симптоме сличне онима код хемизиготних пацијената, иако ови пацијенти имају значајан ниво циркулишућег ензима. Поред тога, акумулација Гб3 у лизозому хемизиготних пацијената почиње у раном детињству или пре рођења, много пре него што се развију клинички релевантни симптоми. Такође не постоји корелација између степена болести и нивоа Гб3 у плазми или урину ни код хемизиготних ни код хетерозиготних пацијената.[29]
Пошто се болест не манифестује у детињству чак ни код пацијената без икакве активности α-галактозидазе А, претпоставља се да акумулација Гб3 није директан узрок Фабријеве болести.
Тренутно се верује да је глоботриаозилсфингозин – метаболит глоботриаозилцерамида – на крају узрок патолошког оштећења код Фабријеве болести. Барем у оштећењу гломерула које доводи до бубрежне инсуфицијенције код Фабријеве болести, лизо-Гб3 игра кључну улогу. Лисо-Гб3 ослобађа ТГФ-β1 и инхибитор макрофага ЦД74 . Патомеханизам који следи подсећа на дијабетесну нефропатију.[30]
Клиничка слика
уреди

Градијентни облици и озбиљност
уредиПрави се разлика између два типа пацијената и прогресивних облика Фабријеве болести: „класичних“ хемизиготних пацијената код којих α-галактозидаза А нема никакву активност, и „атипичних“ хетерозиготних пацијената код којих ензим још увек има резидуалну активност. Класичан ток болести манифестује се у раном појављивању симптома, најчешће у више органа. Насупрот томе, симптоми се код атипичних хетерозиготних пацијената јављају много касније. Поред тога, у таквим случајевима болест може бити локализована, на пример на срчаном мишићу . Мушки пацијенти развијају типичне симптоме Фабријеве болести од детињства. Код жена то често није случај до 40. до 50. године. Због преостале активности α-галактозидазе А, симптоми су често мање изражени.
Симптоми су сложени и могу се разликовати од особе до особе. Рани симптоми су од велике важности за дијагнозу. Већина касних симптома, с друге стране, одређује морталитет (стопу смртности) пацијената.
Индекс озбиљности је број који одређује тежину болести. За пацијенте мушке Фабријеве болести, пенетрантност је оцењена на 100%, а озбиљност на 84%. За пацијенткиње, вредности пенетрације су 70%, а тежине 4%.
Квалитет живота
уредиСимптоми болести узрокују низак квалитет живота, посебно код мушких пацијената. Упоредив је са оним код пацијената са АИДС-ом. Квалитет живота пацијената са Фабријевом болешћу је на сличном нивоу као код пацијената са мултиплом склерозом или реуматоидним артритисом .
Фабријева болест има значајан негативан утицај на психосоцијално окружење оболелих. Више од половине мушких пацијената је неожењено. Велики проценат је незапослених. Депресија је изузетно честа код пацијената са Фабријевом болешћу. Они су недовољно дијагностиковани или се не лече и значајно смањују квалитет живота пацијената. Према једној студији, 46% пацијената има депресију, а 28% има довољно тешку да буде клинички релевантна. Резултати изнад 26 се постижу на Хамилтоновој скали . За разлику од нормалне популације, удео мушкараца са великом депресијом (36%) је већи него код жена (22%).
Неколико студија препоручује психијатријску и неуропсихолошку евалуацију пацијената са Фабријевом болешћу. Детаљно, пацијенти се жале на физичку нелагоду, тугу и психичку патњу . Физички симптоми се повећавају под стресом. Психолошки тестови показују натпросечне поремећаје понашања, неповерење, дефанзивност, емоционална превирања и осећај изолованости. Резултати ових тестова су углавном исти као и код пацијената са болом.
Рани знаци и симптоми
уреди

Бол
уредиЈедан од првих симптома класичног облика Фабријеве болести је бол у рукама и стопалима, акралима . Ове акропарестезије се појављују већ у детињству. Они су узроковани оштећењем танких нервних влакана ( неуропатија малих влакана ) аутономног и периферног соматског нервног система . Око 60 до 80% дечака и девојчица са класичном формом је погођено овим болом.
Пацијенти описују две врсте бола: периодично понављајући пароксизмални напади бола, који се називају и „Фабријеве кризе“, са пекућим болом који се шири из шака и стопала у друге делове тела, и хронични бол, који одговара парестезијама печења и пецкања. Фабријеве кризе могу изазвати грозница, вежба, стрес, исцрпљеност и брзе промене температуре. Ови симптоми се понекад погрешно тумаче као реуматске тегобе, Рејноов синдром , системски еритематозни лупус и, пре свега, болови у расту .
Бол обично нестаје у одраслом добу. Јављају се раније и чешће код дечака него код девојчица. За дечаке, у просеку, са седам година, за девојчице са девет година. Бол има значајан негативан утицај на квалитет живота пацијената.
Гастроинтестинални поремећаји
уредиНелагодност у дигестивном тракту је још један чест, углавном потцењен, рани симптом Фабријеве болести. Ове болести обично трају у одраслом добу. Пацијенти се жале на бол у стомаку , обично након јела, дијареју , мучнину и повраћање . Ово заузврат може бити узрок анорексије (губитак апетита). Ови гастроинтестинални симптоми су вероватно узроковани депозитима Гб3 у аутономним ганглијама (ганглиа аутономица) црева и мезентеричним крвним судовима.
Анхидроза
уредиМноги пацијенти са Фабријевом болешћу не могу да се зноје ( анхидроза ) или то раде само у знатно смањеној мери (хипохидроза). Вредности импедансе коже су стога релативно високе. Анхидроза/хипохидроза може довести до нетолеранције на топлоту и значајних ограничења у спортским активностима код оболелих. У студији на 714 пацијената са Фабријевом болешћу, анхидроза је дијагностикована код 53% мушкараца и 28% жена. Узрок смањене способности знојења су акумулације липида унутар неурона аутономног нервног система.
Ангиокератоми
уредиНајлакше препознатљиви рани симптом Фабријеве болести је ангиокератом. Ово су бенигне црвено-љубичасте лезије коже са благим избочинама. Обично се формирају на задњици, препони, пупку и бутини. Повремено су захваћене и слузокоже, на пример у устима.[32]
У већини случајева, ангиокератоми су мали површински ангиоми који су резултат оштећења васкуларног ендотела коже повезаних са вазодилатацијом у кожи. Са годинама се повећавају у броју и величини и могу се јавити појединачно или у групама.[33]
-
 Хистолошка припрема биопсираног узорка коже пацијента са Фабријевом болешћу. На светлосној микроскопској слици, типичне лезије коже се могу видети као мали површински ангиоми.
Хистолошка припрема биопсираног узорка коже пацијента са Фабријевом болешћу. На светлосној микроскопској слици, типичне лезије коже се могу видети као мали површински ангиоми. -
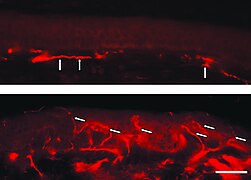 Флуоресцентне слике смрзнутог дела коже пацијента са Фабријевом болешћу. Недостатак интраепидермалних нервних влакана и присуство влакана која припадају субепидермалном нервном плексусу (стрелице) су упадљиви. Доњи узорак коже, с друге стране, долази са леђа пацијента. Овде је упадљива густа инервација епидермиса (стрелице).
Флуоресцентне слике смрзнутог дела коже пацијента са Фабријевом болешћу. Недостатак интраепидермалних нервних влакана и присуство влакана која припадају субепидермалном нервном плексусу (стрелице) су упадљиви. Доњи узорак коже, с друге стране, долази са леђа пацијента. Овде је упадљива густа инервација епидермиса (стрелице). -
 Цртеж након посматрања под микроскопом карминског бојења епидермиса са ангиокератомом, акварел је из чланка Јоханеса Фабрија из 1898. године
Цртеж након посматрања под микроскопом карминског бојења епидермиса са ангиокератомом, акварел је из чланка Јоханеса Фабрија из 1898. године



Вртложна кератопатија
уреди
Карактеристичне замућености рожњаче су најчешћи рани симптом Фабријеве болести. Могу се са сигурношћу дијагностиковати помоћу прорезне лампе и јављају се код скоро свих хемизиготних пацијената. Овај облик замућења рожњаче се назива вертициллата рожњаче или вртложна кератопатија . Двострано је и има карактеристичан узорак крем боје. Облачност не утиче на оштрину вида. Неки лекови, као што су амиодарон и хлорокин , такође изазивају вртложне кератопатије при продуженој употреби.
Касни симптоми
уреди
Оштећење бубрега
уредиКумулативни проценат пацијената са Фабријевом болешћу који имају протеинурију (жута) и хронична бубрежна инсуфицијенција (наранџаста). Црвена крива је стопа смртности. Као и већина симптома Фабријеве болести, оштећење бубрега је прогресивно. Завршава се терминалном инсуфицијенцијом бубрега и узрокује значајно скраћен животни век. У класичној клиничкој слици Фабријеве болести, акумулације Гб3 у ендотелним ћелијама гломерула , у мезангијалним ћелијама , у подоцитима и у ћелијама интерстицијума доводе до оштећења бубрега. Ове ћелије су диференциране епителне ћелије . Такође у епителу Хенлеове петљеАкумулације гликосфинголипида налазе се у бубрежним артериолама и дисталним тубулима и у ендотелијуму и ћелијама глатких мишића артериола . Наслаге Гб3 у цитоплазми могу се јасно видети у трансмисионом електронском микроскопу (ТЕМ). Они су у облику мијелинских структура и наслањају се на ћелијско језгро . Са повећањем акумулације Гб3, мезангијум се шири, што доводи до сегментне или глобалне гломерулосклерозе са задебљањем базалних мембрана . Као могући механизми разматрају се микроваскуларне лезије и оштећења подоцита, који су важни за рад филтера, и епителних ћелија тубула.
У класичном току болести, оштећење бубрега почиње у другој до трећој деценији живота . Прво, може се приметити микроалбуминурија , што представља излучивање малих количина протеина албумина у урину, која се развија у протеинурију (излучивање веће количине протеина урином). Курс је сличан дијабетичкој нефропатији и директно доприноси прогресији Фабри нефропатије. Протеинурија постаје тежа са годинама. Изостенурија се развија како болест напредује, односно бубрези потпуно губе способност концентрације или разблаживања. Ово је праћено променама тубуларне реапсорпције , секреције и излучивања .
У почетку, оштећење бубрега је маскирано гломеруларном хиперфилтрацијом . Међутим, када је критичан број нефрона оштећен, бубрежна функција прогресивно опада. Брзина гломеруларне филтрације , мера филтрационог капацитета бубрега, смањује се за око 12 мл/мин годишње ако се не лечи. У трећој до петој деценији живота, функција бубрега се постепено погоршава и јавља се бубрежна азотемија – ово је абнормално повећање азотних метаболита као што су уреа и креатинин у крви. У овој фази, фиброза , склероза и тубуларна атрофија доминирају Фабријевом нефропатијом и на крају доводе до завршног стадијума бубрежне болести, која се јавља код мушких пацијената у четвртој до петој деценији. Око 17% свих мушкараца и 1% свих пацијената оболелих од Фабријеве болести развије завршну бубрежну инсуфицијенцију и захтева дијализу. Половина пацијената је млађа од 53 године. Више од половине пацијената са Фабријевом болешћу развија нефропатију током болести . Завршна бубрежна болест је главни фактор уМорбидитет и морталитет . Без терапије замене бубрега, уремија (тровање урином) ће неизбежно довести до смрти.
- Узорци ткива из бубрега пацијената са Фабријевом болешћу
-
 Овај светлосни микрограф показује акумулацију Гб3 у гломеруларној ендотелији, мезангијалним ћелијама, интерстицијским ћелијама и подоцитима.
Овај светлосни микрограф показује акумулацију Гб3 у гломеруларној ендотелији, мезангијалним ћелијама, интерстицијским ћелијама и подоцитима. -
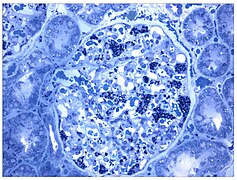 Такође светлосни микрограф. Повећана акумулација Гб3 у подоцитима визуелизована је љубичастом бојом.
Такође светлосни микрограф. Повећана акумулација Гб3 у подоцитима визуелизована је љубичастом бојом. -
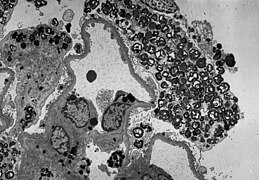 ТЕМ слика показује масивну електрон-густу (= црну) акумулацију гликосфинголипида у лизозому подоцита
ТЕМ слика показује масивну електрон-густу (= црну) акумулацију гликосфинголипида у лизозому подоцита -
 Такође ТЕМ слика. Приказује инклузије гликосфинголипида различитих облика и величина у ћелијама дисталног тубула.
Такође ТЕМ слика. Приказује инклузије гликосфинголипида различитих облика и величина у ћелијама дисталног тубула. -
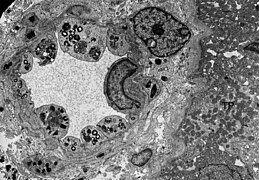 ТЕМ слика ендотелије и глатких мишићних ћелија бубрежне артериоле са инклузијама гликосфинголипида
ТЕМ слика ендотелије и глатких мишићних ћелија бубрежне артериоле са инклузијама гликосфинголипида





Оштећење срца
уреди
Око 40 до 60% људи са Фабријевом болешћу има срчане симптоме као што су хипертрофија леве коморе (задебљање леве коморе срца), абнормални срчани ритам (аритмија), ангина пекторис (бол у грудима сличан нападу) и диспнеја (отежано дисање).[34][35] Срчану аритмију и поремећену варијабилност срчане фреквенције узрокују синусни чвор, проводни систем и неравнотежа између тонуса симпатикуса и парасимпатикуса. Дијастолна дисфункција и хипертрофија леве коморе су важни симптоми Фабријеве болести. Ови симптоми су генерално озбиљнији за мушкарце него за жене. Исхемија миокарда (поремећаји циркулације срчаног мишића) је резултат лошег коронарног протока крви.[36]
У старости се развија прогресивна фиброза миокарда, која је и реверзибилно интерстицијална и иреверзибилно ожиљна (замена фиброзе).[37] У скоро свим случајевима, иреверзибилне ожиљне фиброзе формирају се прво у задњем бочном срчаном зиду иу средњем миокарду. Код пацијената у крајњем стадијуму, трансмурална (обухвата целу дебљину зидног слоја срца) ожиљна фиброза постепено смањује срчану функцију до тачке конгестивне срчане инсуфицијенције.[38] Малигне аритмије су одговорне за већину срчаних смрти код пацијената са Фабријевом болешћу.[34][11][39]
Структурне промене леве коморе у срцу су уобичајене код пацијената са Фабријевом болешћу. Углавном концентричне хипертрофије,[40] могу се учинити видљивим ехокардиографијом (ултразвуком срца) или магнетном резонанцом срца (МРТ). Пошто задњи зид леве коморе срца постаје све тањи и тањи са старењем због замене фиброзе, мерење дебљине септума - то је дебљина преградног зида између леве и десне половине срца - је посебно важно. Без обзира на структурне промене, систола, фаза у којој се крв истискује из леве и десне коморе, изгледа да је у великој мери очувана када се мери конвенционалним методама.[41] Кардиомиопатију узроковану Фабријевом болешћу карактерише смањена контракција и опуштање срчаног мишића. Доплер ткива (и снимање брзине ткива и слика брзине напрезања) може квантификовати функцију срчаног мишића.[11] Овом методом, кардиомиопатија се може дијагностиковати пре него што се развије хипертрофија леве коморе.[42]
- Ехокардиограми пацијената са Фабријевом болешћу
-
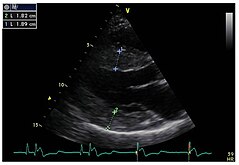 Парастернална дуга оса: Хипертрофија леве коморе са повећаном дебљином септума је јасно видљива.
Парастернална дуга оса: Хипертрофија леве коморе са повећаном дебљином септума је јасно видљива. -
 Парастернална кратка оса: слика такође показује хипертрофију леве коморе.
Парастернална кратка оса: слика такође показује хипертрофију леве коморе. -
 Доплер ехокардиографија митралног прстена (митралног прстена) са скоро нормалном систолном функцијом
Доплер ехокардиографија митралног прстена (митралног прстена) са скоро нормалном систолном функцијом




Код многих болесника са Фабријевом болешћу, десна комора је такође хипертрофична (хипертрофија десне коморе). Комора је нормалне величине и систола је такође нормална; дијастолна функција је значајно смањена. Две трећине пацијената са хипертрофијом десне коморе такође показују симптоме хипертрофија десне коморе.[43]
Хипертрофија десне коморе је вероватан разлог зашто пацијенти са добром функцијом срца леве коморе имају ниску физичку издржљивост и пате од органомегалије (увећање органа) и лимфедема.[11][44]
Због оштећења срчане функције, електрокардиограми (ЕКГ) одраслих пацијената оболелих од Фабријеве болести са класичним обликом болести показују карактеристичне промене.[11]
Цереброваскуларно оштећење
уредиРани симптоми периферних неуропатија, који се обично јављају у адолесценцији, често су праћени у одраслом добу цереброваскуларним обољењима и аутономним дисфункцијама (болести или функционални поремећаји аутономног нервног система). Неке од најнеповољнијих неуролошких карактеристика Фабријеве болести су узроковане церебралним вишеструким (мултифокалним) поремећајима циркулације у малим крвним судовима.[45] Цереброваскуларне промене могу довести до различитих знакова и симптома. Спектар се креће од главобоље и вртоглавице, преко пролазних исхемијских напада и исхемијских можданих удара,[46] до васкуларне деменције.[11][47]
Преваленција церебралног инфаркта је око 6,9% код мушкараца и 4,3% код жена. Значајно је већи него у општој популацији. Средња старост код првог можданог удара је око 39 година за мушкарце са Фабријевом болешћу и 46 за жене. Мождани инфаркт је неретко прва манифестација Фабријеве болести.[45] У већини случајева, инфаркт мозга изазивају мали крвни судови. Поред тога, долихоектазије (син. дилатативне артериопатије, проширење артерија) вертебробазиларне циркулације су такође описане као окидачи.[11] Формирање тромба може бити подстакнуто повећаном адхезијом неутрофилних гранулоцита и моноцита на ендотел или локалним повећаним протоком крви (хиперперфузија).[11][48] Ниво ензима мијелопероксидазе у серуму је биомаркер за ризик од васкулопатије код мушкараца са Фабријевом болешћу.[11]
-
 МРИ аксијалног мозга 27-годишњег пацијента са Фабријевом болешћу са исхемијским можданим ударом који показује мождани удар у левој хемисфери малог мозга. Пацијент није имао других симптома болести
МРИ аксијалног мозга 27-годишњег пацијента са Фабријевом болешћу са исхемијским можданим ударом који показује мождани удар у левој хемисфери малог мозга. Пацијент није имао других симптома болести -
 Хиперинтензитет беле материје , лакунарни церебрални инфаркт и микрокрварење.
Хиперинтензитет беле материје , лакунарни церебрални инфаркт и микрокрварење. -
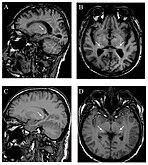 Т1 - пондерисани сагитални (А) и аксијални (Б) МРИ показују симетрично висок сигнал у таламусу (тзв. пулвинарски знак ) 66-годишњег пацијента. (Ц) и (Д), такође Т 1 -пондерисани, показују пулвинарски знак код 42-годишњег пацијента.
Т1 - пондерисани сагитални (А) и аксијални (Б) МРИ показују симетрично висок сигнал у таламусу (тзв. пулвинарски знак ) 66-годишњег пацијента. (Ц) и (Д), такође Т 1 -пондерисани, показују пулвинарски знак код 42-годишњег пацијента. -
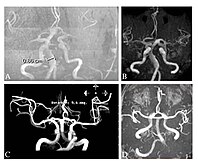 Временска магнетна резонантна ангиографија четири пацијента са Фабријевом болешћу показује проширене (екстатичне) крвне судове (долихоектазија вертебробазиларне циркулације).
Временска магнетна резонантна ангиографија четири пацијента са Фабријевом болешћу показује проширене (екстатичне) крвне судове (долихоектазија вертебробазиларне циркулације).




Остали каснији ефекати болести
уреди

Оштећење бубрега, срца и мозга чини највећи део смртности од Фабријеве болести. Друге касне последице су клинички релевантне, али мало или нимало доприносе морталитету болести. На пример, оштећења органа слуха и равнотеже су широко распрострањена. 80% мушких и 77% женских пацијената показује прогресивни губитак равнотеже.[49] Функција вестибуларног система може се проверити помоћу импулсног теста главе. Код хемизиготних пацијената са класичним током болести, прогресивни губитак слуха и изненадна глувоћа су изузетно чести.[50] Акумулација α-галактозидазе А такође може довести до тинитуса и вртоглавице.[51]

Дисајни путеви су такође погођени болешћу код многих пацијената са Фабријевом болешћу. Отежано дисање (диспнеја), хронични кашаљ, пискање и пискање су уобичајени код оба пола.[52] Према једној студији, 61% мушкараца и 26% жена има опструкције дисајних путева.[11]
Промене на скелету, које суштински утичу на густину костију, такође су чест касни симптом Фабријеве болести. У једној студији, код 88% пацијената просечне старости од 31 године и класичног тока болести дијагностикована је или остеопенија или узнапредовали стадијум, остеопороза, коришћењем дуалне рендгенске апсорпциометрије (ДКСА).[53] У следећој већој студији, остеопенија је пронађена код приближно 50% пацијената са Фабријевом болешћу. Смањена густина костију може довести до спонтаних прелома.[54][55]
Дијагноза
уредиВажност ране дијагнозе
уредиДијагностиковање Фабријеве болести што је раније могуће је важно из више разлога. С једне стране, од 2001. године постоји могућност да се болест лечи узрочно. Квалитет живота оболелих може се значајно побољшати, а оштећење органа барем смањити или одложити. С друге стране, генетска предиспозиција за болест може се препознати код чланова породице пре него што се појаве први симптоми. У таквим случајевима могуће је праћење развоја болести и рана терапија пре него што болест постане симптоматска.[56]
Погрешна дијагноза
уредиЗбог реткости болести, већина педијатара и интерниста погрешно поставља дијагнозу Фабријеве болести и нетачно је лечи. Студија из 2010. анализирала је историју болести 45 пацијената са Фабријевом болешћу. Већина пацијената се у младости жалила на неуропатски бол као први симптом болести - у већини случајева погрешно је дијагностификован као "реуматска грозница". Седам пацијената је годинама лечено пеницилином. Десет пацијената са боловима у стомаку дијагностиковано је тровање храном или "неспецифични бол". Почетном симптому анхидрозе није било могуће приписати узрок и ангиокератоми су интерпретирани као петехије. У просеку је било потребно 19,7 година да се постави тачна дијагноза Фабријеве болести.[57] У претходној студији на 366 пацијената, ова временска разлика је била 13,7 година за мушкарце и 16,3 године за жене.[58] Британска студија из 2001. године пронашла је средњу старост од 22 године за мушке пацијенте у време иницијалне дијагнозе, која је постављена у просечном интервалу од 8 година након првих симптома.[59]
У дугом периоду између првих симптома и тачне дијагнозе, многи пацијенти морају да издрже дугу и фрустрирајућу одисеју од лекара до лекара.[60] У већини случајева тачну дијагнозу поставља случајно офталмолог преко вертициллата рожњаче (вортекс кератопатија) или дерматолог преко ангиокератома.[59]
Тачна дијагноза
уредиУ случају класичног облика Фабријеве болести, клиничка слика може значајно допринети раној исправној дијагнози; посебно ангиокератоми и вортекс кератопатија. Одређивање активности α-галактозидазе А из плазме или из леукоцита помоћу ензимског теста даје дијагностичку сигурност код пацијената мушког пола.[61] Одређивање из плазме понекад може довести до погрешне дијагнозе Фабријеве болести, због чега се препоручује да се налаз провери одређивањем активности леукоцита.[62] Код женских пацијената ова метода је често недовољна. Не успева код више од 30% пацијената са Фабријевом болешћу јер је резидуална активност ензима превисока за тест. [63] Стога, свим пацијентима за које се сумња да имају Фабријеву болест треба дијагностиковати генотипизацију.[18]
Ген је секвенциониран и упарен са познатим ГЛА мутацијама. Поред тога, корисно је одређивање биомаркера Лисо-Гб3. Ако је активност ензима нејасна, вредност Лисо-Гб3 већа од 1,3 нмол/л може указивати на Фабријеву болест код људи са неспецифичним симптомима Фабријеве болести (ЛВХ или ЦКД, итд.).[64] Тест осушене крви (ДБС) који се лако може интегрисати у свакодневну праксу сада је доступан за мерење активности ензима и Лисо-Гб3, као и за генетску анализу: неколико капи крви ставља се на картицу осушене крви. Након што се осуше, картица се шаље поштом у специјализовану лабораторију. Тамо се крв уклања са филтер картице и припрема за даља испитивања. Лизо-ГЛ-3 је добар маркер да се са сигурношћу искључи класична Фабријева болест. Код особа са неизвесним ГЛА значајем, које показују неспецифичне симптоме Фабријеве болести (ЛВХ или ЦКД, итд.) и немају карактеристичне, фенотипске или биохемијске карактеристике класичне Фабријеве болести, треба користити > 1,3 нмол/Л мислити на Фабријеву болест.[64]
У принципу, нивои Гб3 у плазми и урину,[29] такође се могу користити за дијагнозу.[65] Вредности урина су поузданије од вредности у плазми у погледу њихове информативне вредности код мушких и женских пацијената; међутим, неки пацијенти са касним стадијумом Фабријеве болести или са одређеним мутацијама (као што је п.Асн215Сер) имају нормалне концентрације Гб3 у урину.[11]
Кобов синдром се мора разликовати у диференцијалној дијагнози.
Пренатална дијагноза
уредиДијагноза Фабријеве болести је пренатална, односно пренатална. За ово се може користити биохемијска или молекуларна пренатална дијагностика. У првом случају, активност α-галактозидазе А хорионских ресица се може мерити директно или у ћелијској култури. Уклањање узорковањем хорионских ресица могуће је у десетој недељи трудноће. Дијагноза је могућа око 14. недеље трудноће из култивисаних амнионских ћелија (ћелија у амнионској течности), које се из плодове воде уклањају амниоцентезом. Одређивање генотипа помоћу ДНК анализе (генски тест) је компликованије. Генетско саветовање се обично обавља пре пренаталне дијагнозе. Из етичких разлога, пренатална дијагноза Фабријеве болести, посебно код женских фетуса, веома је контроверзна. Са доступношћу терапије замене ензима, ова дискусија се проширила на мушке фетусе. Неки аутори генерално препоручују пренаталну дијагнозу само за мушке фетусе. Пол фетуса се може одредити из крви мајке у 9. до 11. недељи трудноће.[11]
У принципу, преимплантациона дијагностика је могућа и већ је спроведена. До сада (од септембра 2011.) није било публикација о томе.[66]
Скрининг новорођенчета
уредиМорбус Фабри тренутно није део пројекција новорођенчади у Немачкој и Аустрији. Претпоставка је да скрининг за одређену урођену болест има смисла само ако постоји и опција лечења за њу. Са доступношћу терапије замене ензима, ова премиса код Фабријеве болести више не важи.[67] ХПЛЦ-МС се може користити за брзу и релативно јефтину анализу неколико лизозомалних болести складиштења од сувих крвних мрља (ДБС).[68] Тренутно је у току неколико великих студија како би се потврдила поузданост методе.[68] Лизо-ГЛ-3 плазме се може лако мерити тестом суве крви и нуди ефикасан и специфичан биомаркер за скрининг новорођенчади за које се сумња да имају Фабријеву болест пре него што се појаве први симптоми.[69]
Терапија
уреди
Као и код свих болести лизозомског складиштења, развој ефикасних метода лечења, посебно активних састојака, је изузетно тежак, па тако и за Фабријеву болест. С једне стране, због ниске инциденце, постоји врло мали број пацијената за спровођење клиничких студија, а са друге стране, захтеви у погледу безбедности лека када се узимају током дужег временског периода су веома високи. Лек треба узимати доживотно, а идеално пре него што се појаве први симптоми, односно углавном здрави пацијенти.[70] Због реткости болести, тржиште за развијени лек је изузетно мало. Високи трошкови развоја који су уобичајени у фармацеутској индустрији се тако распоређују на мали број пацијената, што резултује веома високим трошковима лечења по пацијенту.
До 2001. године пацијенти са Фабријевом болешћу могли су се лечити само симптоматски или палијативно . До ове тачке, третман се у суштини састојао од избегавања стимуланса који изазивају бол као што су стрес, физички напор, топлота, сунчева светлост и оштре промене температуре. За ублажавање бола коришћене су високе дозе аналгетика . Анхидроза је сузбијана повећањем уноса течности по топлом времену и избегавањем физичког напора. Дијета са мало масти и лекови су коришћени за ублажавање гастроинтестиналних симптома. дијета за бубрегеје прописан за благу протеинурију. Антикоагуланси су прописани за спречавање можданог удара. Завршна бубрежна инсуфицијенција је – као што је и данас случај – лечена терапијом замене бубрега (дијализа или трансплантација бубрега).[71]
Терапија замене ензима
уредиЕнзимска супституциона терапија (ЕРТ) је тренутно (од септембра 2011.) једина могућност за узрочни третман ( каузална терапија ) Фабријеве болести.[72] За пацијенте са Фабријевом болешћу у Европској унији постоје два лека који се користе за лечење болести: агалзидаза алфа и агалзидаза бета. Обе су биотехнолошки произведене варијанте α-галактозидазе А. Агалсидаза бета је доступна пацијентима у САД од априла 2003. године. До данас (од септембра 2011.), агалсидаза алфа није одобрена у Сједињеним Америчким Државама. Осим у 27 земаља ЕУ, одобрен је у укупно 45 земаља, укључујући Канаду, Јапан, Бразил и Кину (од маја 2011).[73]
Оба лека су генетски модификована. Док ћелијска линија хуманих фибробласта производи ензим за агалзидазу алфа, ћелије јајника кинеског хрчка ( ЦХО) се користе за агалзидазу бета.[74] Агалсидаза бета је химерни протеин. Два ензима су идентична у својој секвенци аминокиселина, али се незнатно разликују по типу гликозилације због различитог облика експресије протеина током производње . Главне разлике су у пропорцији сијаличних киселинаи маноза-6-фосфат. Агалсидаза бета има већи удео потпуно сијалированих олигосахарида и виши степен фосфорилације. У ин витро експериментима, повећано везивање за маноза-6-фосфатни рецептор и веће узимање у Фабри фибробластима могло се утврдити за агалзидазу бета.[75] Насупрот томе, никаква функционална разлика између два ензима се не може идентификовати ин виво . Такође се не разликују у погледу степена унакрсне реактивности антитела . Оба ензима се морају применити интравенозно. Ензими који би требало да имају системски ефекат у основи нису доступни орално, јер се у цревима у великој мери разлажу на своје компоненте (аминокиселине). Агалсидаза алфа се примењује током 40 минута у дози од 0,2 мг/кг телесне тежине сваке две недеље . Агалсидаза бета се примењује истом брзином.[72] Међутим, доза је 1 мг/кг телесне тежине, а трајање инфузије је у почетку четири сата. Ово време инфузије се онда може смањити на 90 минута ако се добро подноси.
Ефикасност
уредиОсип код 39-годишњег пацијента након инфузије α-галактозидазе А. Пацијент је развио имуноглобулин Е против агалзидазе бета. Упркос примени оралних кортикостероида , парацетамола и хидроксизина , током фаза инфузије су се развили опсежни осип на кожи и бронхоспазам . Функција бубрега је наставила да се погоршава. Због тога је терапија замене ензима прекинута. Пацијент има мутацију п.Ала121Про која се не може лечити испитиваним мигаластатом. У време пријема, разговарало се о комбинацији имуносупресије и терапије замене ензима ради даљег лечења. Због спорог напредовања Фабријеве болести током деценија, као и због њене реткости, постоји само неколико поузданих података о дугорочним успесима лечења. У компаративној студији у периоду од 24 месеца, није пронађена разлика између агалзидазе алфа и агалзидазе бета у погледу основних мерљивих параметара болести. Нарочито у ендотелним ћелијама, терапија замене ензима може значајно да смањи акумулацију Гб3 у лизозому. Ово је од великог значаја за лечење примарних фактора морбидитета (бубрежна инсуфицијенција, срчана и цереброваскуларна обољења). Међутим, ефикасност терапије код пацијената са узнапредовалим симптомима је прилично скромна. Због тога је што раније могуће лечење посебно важно за успех терапије. У терапији замене ензима, деградација Гб3 зависи од типа ћелије. У бубрезима, Гб3 се такође разграђује у мезангијалним ћелијама гломерула и ћелијама у интерстицијуму бубрежног кортекса, поред васкуларне ендотелије. Смањење Гб3 у глатким мишићима артериола и малих артерија, подоцитима и у епителу дисталног тубула је значајно лошије.
У клиничким испитивањима, примена агалзидазе алфа је смањила бол и смањила хипертрофију леве коморе. Стабилизирана је функција бубрега, побољшан слух и способност знојења. Све у свему, квалитет живота је значајно побољшан. Код пацијената са хроничном бубрежном инсуфицијенцијом, прогресија до завршног стадијума бубрежне инсуфицијенције може бити одложена. Током третмана агалзидазом бета, детектовано је смањење Гб3 у различитим ћелијама. Такође нема поновне акумулације Гб3. Примена агалзидазе бета значајно смањује ризик од озбиљног клиничког догађаја (нпр. инфаркт миокарда, завршна бубрежна болест или смрт).[76]
Нежељени ефекти
уреди
Отприлике половина свих пацијената доживљава благе до умерене реакције повезане са инфузијом, које достижу врхунац између пете и осме инфузије. Поред тога, неки пацијенти показују грозницу и мрзлицу. Ови нежељени ефекти су краткотрајни, нису озбиљни и могу се лечити конзервативно. После три до пет година лечења, само 10 до 20% пацијената и даље показује реакције повезане са инфузијом, при чему се очигледно развија толеранција на инфузију. Још увек није познат тачан узрок реакција повезаних са инфузијом.
Постоји сумња на специфично ИгГ антитело-Едукација о инфузираном ензиму. ИгГ сероконверзија је откривена код 24% пацијената који су примали агалсидазу алфа и 88% пацијената који су примали агалсидазу бета. До формирања антитела може доћи код пацијената који немају никакву резидуалну активност α-галактозидазе. α-галактозидаза је "нова" и "ванземаљска" њиховом имунолошком систему. Формирање антитела против два препарата агалзидазе директно смањује њихову ефикасност.[78] Међутим, студија са више центара је показала да је „прекомерно прскање“ антитела одговарајућим количинама ензима резултирало континуираним ниским нивоима маркера болести Лисо-Гб3.[79]
Трошкови терапије
уредиТрошкови лечења у Немачкој су око 250.000 евра по пацијенту годишње, без обзира да ли се користили агалзидазу алфа или бета.[80] Пошто је заменска терапија ензимима, тренутно једина узрочна терапијска опција за Фабријеву болст, трошкове за то надокнађују обавезна здравствена осигуравајућа друштва у Немачкој и не рачунају се у буџет лекарске ординације за рецепт.[81]
Ампула која садржи 3,5 мг агалзидазе алфа, за дозу од 0,2 мг по кг телесне тежине, кошта 1.685 евра у Француској. За ампулу са 35 мг агалзидазе бета, са дозом од 1 мг по кг телесне тежине, наплаћује се 3.370 €. Ово резултује идентичним годишњим трошковима лечења по пацијенту, који у Француској износе 161.781 € за пацијента тежине 70 кг (2009. године).[11]
Истовремене терапије
уредиНарочито код младих пацијената, терапија замене ензима је прилично успешна у смањењу неуропатског бола.[82] Међутим, у многим случајевима је индикована истовремена терапија бола . карбамазепином,[83] ако је потребно у комбинацији са прегабалином,[84] препоручује се као лек првог избора за лечење неуропатског бола код Фабријеве болести .
У случају неподношљивих болних криза, могу се користити и опиоиди.[85] Лекови као што је метоклопрамид се користе за лечење гастроинтестиналних симптома препоручује се за сузбијање поремећаја кретања у горњем делу гастроинтестиналног тракта (поремећаји мотилитета).[86]
Ако постоји повећано излучивање протеина у урину као индикација оштећења бубрега, додатно лечење АЦЕ инхибиторима или АТ1 антагонистима , две сродне класе антихипертензивних лекова, може одложити напредовање оштећења бубрега.[87] Ако је срце већ оштећено, АЦЕ инхибитори могу смањити артеријски васкуларни отпор, а тиме и артеријски крвни притисак. Ово такође смањује предоптерећење и накнадно оптерећење срца у случају миокардне инсуфицијенције и повећава минутни волумен срца . Број откуцаја срца и потреба миокарда за кисеоником могу се смањити бета-блокаторима. Срчане аритмије се могу кориговати нпр. амиодароном. Поред тога, још увек постоје хируршке мере, као што су имплантација пејсмејкера , коронарног стента , вештачког срчаног залиска или премоснице коронарне артерије.[88]
Извори
уреди- ^ а б в г д Zarate, Yuri A.; Hopkin, Robert J. (2008-10-18). „Fabry's disease”. The Lancet (на језику: енглески). 372 (9647): 1427—1435. ISSN 0140-6736. PMID 18940466. S2CID 21137862. doi:10.1016/S0140-6736(08)61589-5.
- ^ Warnock, D. G.; West, M. L. (2006). „Diagnosis and management of kidney involvement in Fabry disease”. Adv Chronic Kidney Dis. 13 (2): 138—147. PMID 16580615. doi:10.1053/j.ackd.2006.01.013.
- ^ а б MacDermot, K. D.; Holmes, A.; Miners, A. H. (1. 11. 2001). „Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females”. Journal of Medical Genetics. 38 (11): 769—775. ISSN 1468-6244. PMC 1734754
 . PMID 11732485. doi:10.1136/jmg.38.11.769.
. PMID 11732485. doi:10.1136/jmg.38.11.769.
- ^ Fabry, H. (2001). „An historical overview of Fabry disease”. Journal of Inherited Metabolic Disease. 24: 3—7. PMID 11758677. S2CID 38947917. doi:10.1023/A:1012443001449.
- ^ Mehta, A.; Beck, M.; Eyskens, F.; Feliciani, C.; Kantola, I.; Ramaswami, U.; Rolfs, A.; Rivera, A.; Waldek, S.; Germain, D. P. (2010). „Fabry disease: A review of current management strategies”. QJM. 103 (9): 641—59. PMID 20660166. doi:10.1093/qjmed/hcq117..
- ^ Desnick, R. J.; Brady, R.; Barranger, J.; Collins, A. J.; Germain, D. P.; Goldman, M.; Grabowski, G.; Packman, S.; Wilcox, W. R. (2003). „Fabry disease, an under-recognized multisystemic disorder: Expert recommendations for diagnosis, management, and enzyme replacement therapy”. Annals of Internal Medicine. 138 (4): 338—46. PMID 12585833. S2CID 12227778. doi:10.7326/0003-4819-138-4-200302180-00014.
- ^ Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R. J. (2006). „High incidence of later-onset Fabry disease revealed by newborn screening”. Am J Hum Genet. 79 (1): 31—40. PMC 1474133 . PMID 16773563. doi:10.1086/504601..
- ^ а б Gold, K. F.; Pastores, G. M.; Botteman, M. F.; Yeh, J. M.; Sweeney, S.; Aliski, W.; Pashos, C. L. (2002). „Quality of life of patients with Fabry disease”. Quality of Life Research : An International Journal of Quality of Life Aspects of Treatment, Care and Rehabilitation. 11 (4): 317—327. PMID 12086117. S2CID 25530468. doi:10.1023/a:1015511908710.
- ^ C. Vetter: Repetitorium: Morbus Fabry. (PDF) In: ZM. Band 101, Nr. 1A, vom 1. Januar 2011, S. 41–45.
- ^ Hwu, W. L.; Chien, Y. H.; Lee, N. C.; Chiang, S. C.; Dobrovolny, R.; Huang, A. C.; Yeh, H. Y.; Chao, M. C.; Lin, S. J.; Kitagawa, T.; Desnick, R. J.; Hsu, L. W. (2009). „Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A)”. Human Mutation. 30 (10): 1397—1405. PMC 2769558 . PMID 19621417. doi:10.1002/humu.21074., 2009, S. 1397–1405
- ^ а б в г д ђ е ж з и ј к л љ м н Germain, Dominique P. (2010). „Fabry disease”. Orphanet Journal of Rare Diseases. 5: 30. PMC 3009617 . PMID 21092187. doi:10.1186/1750-1172-5-30 .
- ^ Lin, Hsiang-Yu; Chong, Kah-Wai; Hsu, Ju-Hui; Yu, Hsiao-Chi; Shih, Chun-Che; Huang, Cheng-Hung; Lin, Shing-Jong; Chen, Chen-Huan; Chiang, Chuan-Chi; Ho, Huey-Jane; Lee, Pi-Chang; Kao, Chuan-Hong; Cheng, Kang-Hsiang; Hsueh, Chuen; Niu, Dau-Ming (2009). „High Incidence of the Cardiac Variant of Fabry Disease Revealed by Newborn Screening in the Taiwan Chinese Population”. Circulation: Cardiovascular Genetics. 2 (5): 450—456. PMID 20031620. S2CID 10456593. doi:10.1161/CIRCGENETICS.109.862920..
- ^ Gaspar, Paulo; Herrera, Julio; Rodrigues, Daniel; Cerezo, Sebastián; Delgado, Rodrigo; Andrade, Carlos F.; Forascepi, Ramón; MacIas, Juan; Del Pino, Maria D.; Prados, Maria D.; De Alegria, Pilar R.; Torres, Gerardo; Vidau, Pedro; Sá-Miranda, Maria C. (2010). „Frequency of Fabry disease in male and female haemodialysis patients in Spain”. BMC Medical Genetics. 11: 19. PMC 2837018 . PMID 20122163. doi:10.1186/1471-2350-11-19 .
- ^ Nowak, A.; Mechtler, T. P.; Hornemann, T.; Gawinecka, J.; Theswet, E.; Hilz, M. J.; Kasper, D. C. (2018). „Genotype, phenotype and disease severity reflected by serum LysoGb3 levels in patients with Fabry disease” (PDF). Mol Genet Metab. 123 (2): 148—153. PMID 28728877. doi:10.1016/j.ymgme.2017.07.002.
- ^ „OMIM Gene/Loci: 413 – 422 of 718 on Chromosome X”. www.omim.org. Приступљено 2022-02-23.
- ^ „Fabry Disease”. The Medical Biochemistry Page (на језику: енглески). 2020-05-15. Приступљено 2022-02-23.
- ^ Pinto, Louise LC; Vieira, Taiane A.; Giugliani, Roberto; Schwartz, Ida VD (2010). „Expression of the disease on female carriers of X-linked lysosomal disorders: A brief review”. Orphanet Journal of Rare Diseases. 5: 14. PMC 2889886 . PMID 20509947. doi:10.1186/1750-1172-5-14 .
- ^ а б Germain, D.P.; Benistan, K.; Angelova, L. (2010). „X-linked inheritance and its implication in the diagnosis and management of female patients in Fabry disease”. La Revue de Médecine Interne. 31: S209—S213. PMID 21211665. doi:10.1016/S0248-8663(10)70013-8..
- ^ Dobyns, William B.; Filauro, Allison; Tomson, Brett N.; Chan, April S.; Ho, Allen W.; Ting, Nicholas T.; Oosterwijk, Jan C.; Ober, Carole (2004). „Inheritance of most X-linked traits is not dominant or recessive, just X-linked”. American Journal of Medical Genetics. 129A (2): 136—143. PMID 15316978. S2CID 42108591. doi:10.1002/ajmg.a.30123.
- ^ Migeon, Barbara R. (2008). „X Inactivation, Female Mosaicism, and Sex Differences in Renal Diseases”. Journal of the American Society of Nephrology. 19 (11): 2052—2059. PMID 18448583. doi:10.1681/ASN.2008020198.. (Review).
- ^ Kobayashi, M.; Ohashi, T.; Sakuma, M.; Ida, H.; Eto, Y. (2008). „Clinical manifestations and natural history of Japanese heterozygous females with Fabry disease”. Journal of Inherited Metabolic Disease. 31: 483—487. PMID 18202903. S2CID 32234710. doi:10.1007/s10545-007-0740-6..
- ^ Redonnet-Vernhet, I; Ploos van Amstel, J K; Jansen, R P; Wevers, R A; Salvayre, R; Levade, T (1996). „Uneven X inactivation in a female monozygotic twin pair with Fabry disease and discordant expression of a novel mutation in the alpha-galactosidase A gene.”. Journal of Medical Genetics. 33 (8): 682—688. ISSN 0022-2593. PMC 1050704 . PMID 8863162. doi:10.1136/jmg.33.8.682.
- ^ Maier, Esther M.; Osterrieder, Stephanie; Whybra, Catharina; Ries, Markus; Gal, Andreas; Beck, Michael; Roscher, Adelbert A.; Muntau, Ania C. (2007). „Disease manifestations and X inactivation in heterozygous females with Fabry disease”. Acta Paediatrica. 95 (451): 30—38. PMID 16720462. S2CID 15437239. doi:10.1111/j.1651-2227.2006.tb02386.x..
- ^ а б в Mehta, A.; Beck, M.; Sunder-Plassmann, G.; Gal, A.; Schäfer, E.; Rohard, I. (2006). „The genetic basis of Fabry disease”. PMID 21290673.
- ^ A. Gal: Molecular genetics of Fabry disease and Genotype-phenotype correlation. In: D. Elstein, G. Altarescu, M. Beck (eds.): Fabry disease. Verlag Springer. Elstein, Deborah; Altarescu, Gheona; Beck, Michael (2010). Fabry Disease. Springer. стр. 3—19. ISBN 978-90-481-9032-4..
- ^ R. O. Brady: Fabry Disease – An Overview. In: D. Elstein, G. Altarescu, M. Beck (eds.): Fabry Disease. Verlag Springer. Elstein, Deborah; Altarescu, Gheona; Beck, Michael (2010). Fabry Disease. Springer. ISBN 978-90-481-9032-4.
- ^ Futerman, Anthony H.; Van Meer, Gerrit (2004). „The cell biology of lysosomal storage disorders”. Nature Reviews Molecular Cell Biology. 5 (7): 554—565. PMID 15232573. S2CID 21605400. doi:10.1038/nrm1423. hdl:1874/14471.
- ^ Vedder, Anouk C.; Linthorst, Gabor E.; Houge, Gunnar; Groener, Johannna E.M.; Ormel, Els E.; Bouma, Berto J.; Aerts, Johannes M.F.G.; Hirth, Asle; Hollak, Carla E.M. (2007). „Treatment of Fabry Disease: Outcome of a Comparative Trial with Agalsidase Alfa or Beta at a Dose of 0.2 mg/Kg”. PLOS ONE. 2 (7): e598. Bibcode:2007PLoSO...2..598V. PMC 1913555 . PMID 17622343. doi:10.1371/journal.pone.0000598 .
- ^ а б Vedder, A. C.; Linthorst, G. E.; Van Breemen, M. J.; Groener, J. E. M.; Bemelman, F. J.; Strijland, A.; Mannens, M. M. A. M.; Aerts, J. M. F. G.; Hollak, C. E. M. (2007). „The Dutch Fabry cohort: Diversity of clinical manifestations and Gb3 levels”. Journal of Inherited Metabolic Disease. 30 (1): 68—78. PMID 17206462. S2CID 10333874. doi:10.1007/s10545-006-0484-8..
- ^ Sanchez-Nino, M. D.; Sanz, A. B.; Carrasco, S.; Saleem, M. A.; Mathieson, P. W.; Valdivielso, J. M.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. (2011). „Globotriaosylsphingosine actions on human glomerular podocytes: Implications for Fabry nephropathy”. Nephrology Dialysis Transplantation. 26 (6): 1797—1802. PMID 20504837. doi:10.1093/ndt/gfq306..
- ^ A. P. Burlina, K. B. Sims u. a.: Early diagnosis of peripheral nervous system involvement in Fabry disease and treatment of neuropathic pain: the report of an expert panel. In: BMC neurology. Band 11, 2011, S. 61
- ^ „Angiokeratom • LekarInfo” (на језику: српски). 2014-08-26. Приступљено 2023-02-07.
- ^ R Trickett and H Dowd (октобар 2006). „Angiokeratoma of the scrotum: a case of scrotal bleeding”. Emergency Medicine Journal. 23 (10).e57.
- ^ а б Shah, Jaymin S.; Hughes, Derralynn A.; Sachdev, Bhavesh; Tome, Maite; Ward, Deirdre; Lee, Philip; Mehta, Atul B.; Elliott, Perry M. (2005). „Prevalence and Clinical Significance of Cardiac Arrhythmia in Anderson-Fabry Disease”. The American Journal of Cardiology. 96 (6): 842—846. PMID 16169374. doi:10.1016/j.amjcard.2005.05.033..
- ^ Senechal, M.; Germain, D. P. (2003). „Fabry disease: A functional and anatomical study of cardiac manifestations in 20 hemizygous male patients”. Clinical Genetics. 63 (1): 46—52. PMID 12519371. S2CID 11602510. doi:10.1034/j.1399-0004.2003.630107.x.
- ^ Elliott, P. M.; Kindler, H.; Shah, J. S.; Sachdev, B.; Rimoldi, O. E.; Thaman, R.; Tome, M. T.; McKenna, W. J.; Lee, P.; Camici, P. G. (2005). „Coronary microvascular dysfunction in male patients with Anderson-Fabry disease and the effect of treatment with galactosidase A”. Heart. 92 (3): 357—360. PMC 1860797 . PMID 16085718. doi:10.1136/hrt.2004.054015.
- ^ Moon, J. C.; Sheppard, M.; Reed, E.; Lee, P.; Elliott, P. M.; Pennell, D. J. (2006). „The histological basis of late gadolinium enhancement cardiovascular magnetic resonance in a patient with Anderson-Fabry disease”. Journal of Cardiovascular Magnetic Resonance : Official Journal of the Society for Cardiovascular Magnetic Resonance. 8 (3): 479—482. PMID 16755835. doi:10.1080/10976640600605002.
- ^ Linhart, A.; Kampmann, C.; Zamorano, J. L.; Sunder-Plassmann, G.; Beck, M.; Mehta, A.; Elliott, P. M.; European FOS Investigators (2007). „Cardiac manifestations of Anderson-Fabry disease: Results from the international Fabry outcome survey”. European Heart Journal. 28 (10): 1228—1235. PMID 17483538. doi:10.1093/eurheartj/ehm153..
- ^ Takenaka, Toshihiro; Teraguchi, Hiroyuki; Yoshida, Aichi; Taguchi, Syuhei; Ninomiya, Kenjiro; Umekita, Yoshihisa; Yoshida, Hiroki; Horinouchi, Michiko; Tabata, Kazuhiro; Yonezawa, Suguru; Yoshimitsu, Makoto; Higuchi, Koji; Nakao, Shoichiro; Anan, Ryuichiro; Minagoe, Shinichi; Tei, Chuwa (2008). „Terminal stage cardiac findings in patients with cardiac Fabry disease: An electrocardiographic, echocardiographic, and autopsy study”. Journal of Cardiology. 51 (1): 50—59. PMID 18522775. doi:10.1016/j.jjcc.2007.12.001..
- ^ Linhart, Aleš; Paleček, Tomáš; Bultas, Jan; Ferguson, James J.; Hrudová, Jana; Karetová, Debora; Zeman, Jiři; Ledvinová, Jana; Poupětová, Helena; Elleder, Milan; Aschermann, Michael (2000). „New insights in cardiac structural changes in patients with Fabry's disease”. American Heart Journal. 139 (6): 1101—1108. PMID 10827394. doi:10.1067/mhj.2000.105105..
- ^ Pieroni, Maurizio; Chimenti, Cristina; Ricci, Roberta; Sale, Patrizio; Russo, Matteo Antonio; Frustaci, Andrea (2003). „Early Detection of Fabry Cardiomyopathy by Tissue Doppler Imaging”. Circulation. 107 (15): 1978—1984. PMID 12668521. S2CID 2902358. doi:10.1161/01.CIR.0000061952.27445.A0..
- ^ Pieroni, M.; Chimenti, C.; Russo, A.; Russo, M. A.; Maseri, A.; Frustaci, A. (2004). „Tissue Doppler imaging in Fabry disease”. Current Opinion in Cardiology. 19 (5): 452—457. PMID 15316452. S2CID 20093987. doi:10.1097/01.hco.0000131534.25034.43.
- ^ Niemann, M.; Breunig, F.; Beer, M.; Herrmann, S.; Strotmann, J.; Hu, K.; Emmert, A.; Voelker, W.; Ertl, G.; Wanner, C.; Weidemann, F. (2010). „The right ventricle in Fabry disease: Natural history and impact of enzyme replacement therapy”. Heart. 96 (23): 1915—1919. PMID 20965976. S2CID 2837188. doi:10.1136/hrt.2010.204586..
- ^ Kampmann, C.; Baehner, F. A.; Whybra, C.; Bajbouj, M.; Baron, K.; Knuf, M.; Wiethoff, C. M.; Trübel, H.; Beck, M. (2005). „The right ventricle in Fabry disease”. Acta Paediatrica (Oslo, Norway : 1992). Supplement. 94 (447): 15—8; discussion 9—10. PMID 15895706. S2CID 24554620. doi:10.1111/j.1651-2227.2005.tb02104.x.
- ^ а б Sims, Katherine; Politei, Juan; Banikazemi, Maryam; Lee, Philip (2009). „Stroke in Fabry Disease Frequently Occurs Before Diagnosis and in the Absence of Other Clinical Events”. Stroke. 40 (3): 788—794. PMID 19150871. S2CID 9265154. doi:10.1161/STROKEAHA.108.526293..
- ^ Fellgiebel, Andreas; Müller, Matthias J.; Ginsberg, Lionel (2006). „CNS manifestations of Fabry's disease”. The Lancet Neurology. 5 (9): 791—795. PMID 16914407. S2CID 38935531. doi:10.1016/S1474-4422(06)70548-8..
- ^ Magage, S.; Lubanda, J.‐C.; Susa, Z.; Bultas, J.; Karetová, D.; Dobrovolný, R.; Hřebíček, M.; Germain, D. P.; Linhart, A. (2007). „Natural history of the respiratory involvement in Anderson–Fabry disease”. Journal of Inherited Metabolic Disease. 30 (5): 790—799. PMID 17619837. S2CID 11621381. doi:10.1007/s10545-007-0616-9..
- ^ Degraba, T.; Azhar, S.; Dignat-George, F.; Brown, E.; Boutière, B.; Altarescu, G.; McCarron, R.; Schiffmann, R. (2000). „Profile of endothelial and leukocyte activation in Fabry patients”. Annals of Neurology. 47 (2): 229—233. PMID 10665494. S2CID 19202089. doi:10.1002/1531-8249(200002)47:2<229::AID-ANA13>3.0.CO;2-T.
- ^ Palla, A.; Hegemann, S.; Widmer, U.; Straumann, D. (2007). „Vestibular and auditory deficits in Fabry disease and their response to enzyme replacement therapy” (PDF). Journal of Neurology. 254 (10): 1433—1442. PMID 17934877. S2CID 139765. doi:10.1007/s00415-007-0575-y..
- ^ Sakurai, Yuika; Kojima, Hiromi; Shiwa, Masanori; Ohashi, Toya; Eto, Yoshikatsu; Moriyama, Hiroshi (2009). „The hearing status in 12 female and 15 male Japanese Fabry patients”. Auris Nasus Larynx. 36 (6): 627—632. PMID 19261412. doi:10.1016/j.anl.2009.01.001..
- ^ Germain, D. P.; Avan, P.; Chassaing, A.; Bonfils, P. (2002). „Patients affected with Fabry disease have an increased incidence of progressive hearing loss and sudden deafness: An investigation of twenty-two hemizygous male patients”. BMC Medical Genetics. 3: 10. PMC 134464 . PMID 12377100. doi:10.1186/1471-2350-3-10 .
- ^ Rosenberg, D. M.; Ferrans, V. J.; Fulmer, J. D.; Line, B. R.; Barranger, J. A.; Brady, R. O.; Crystal, R. G. (1980). „Chronic airflow obstruction in Fabry's disease”. The American Journal of Medicine. 68 (6): 898—905. PMID 6247911. doi:10.1016/0002-9343(80)90224-7.
- ^ Germain, D. P.; Benistan, K.; Boutouyrie, P.; Mutschler, C. (2005). „Osteopenia and osteoporosis: Previously unrecognized manifestations of Fabry disease”. Clinical Genetics. 68 (1): 93—95. PMID 15952993. S2CID 40653911. doi:10.1111/j.1399-0004.2005.00457.x..
- ^ Mersebach, Henriette; Johansson, Jan-Ove; Rasmussen, åse Krogh; Bengtsson, Bengt-Åke; Rosenberg, Kirsten; Hasholt, Lis; Sørensen, Sven Asger; Sørensen, Søren Schwartz; Feldt-Rasmussen, Ulla (2007). „Osteopenia: A common aspect of Fabry disease. Predictors of bone mineral density”. Genetics in Medicine. 9 (12): 812—818. PMID 18091430. S2CID 22998638. doi:10.1097/GIM.0b013e31815cb197..
- ^ R. O. Brady: Bone and muscle involvement in Fabry disease. In: D. Elstein, G. Altarescu, M. Beck (eds.): Fabry Disease. Verlag Springer. Elstein, Deborah; Altarescu, Gheona; Beck, Michael (2010). Fabry Disease. Springer. стр. 293—298. ISBN 978-90-481-9032-4.eingeschränkte Vorschau in der Google-Buchsuche
- ^ Grau, A. J.; Schwaninger, M.; Goebel, H. H.; Beck, M. (2003). „Morbus Fabry”. Der Nervenarzt. 74 (6): 489—496. PMID 12799787. S2CID 41809661. doi:10.1007/s00115-003-1513-6..
- ^ Marchesoni, Cintia L.; Roa, Norma; Pardal, Ana María; Neumann, Pablo; Cáceres, Guillermo; Martínez, Pablo; Kisinovsky, Isaac; Bianchi, Silvia; Tarabuso, Ana Lía; Reisin, Ricardo C. (2010). „Misdiagnosis in Fabry Disease”. The Journal of Pediatrics. 156 (5): 828—831. PMID 20385321. doi:10.1016/j.jpeds.2010.02.012..
- ^ Mehta, A.; Ricci, R.; Widmer, U.; Dehout, F.; Garcia De Lorenzo, A.; Kampmann, C.; Linhart, A.; Sunder-Plassmann, G.; Ries, M.; Beck, M. (2004). „Fabry disease defined: Baseline clinical manifestations of 366 patients in the Fabry Outcome Survey”. European Journal of Clinical Investigation. 34 (3): 236—242. PMID 15025684. S2CID 40805388. doi:10.1111/j.1365-2362.2004.01309.x..
- ^ а б MacDermot, K. D.; Holmes, A.; Miners, A. H. (2001). „Anderson-Fabry disease: Clinical manifestations and impact of disease in a cohort of 98 hemizygous males”. Journal of Medical Genetics. 38 (11): 750—760. PMC 173476 . PMID 11694547. doi:10.1136/jmg.38.11.750.
- ^ „Morbus Fabry ist selten, die Symptomatik unspezifisch - Vor der Diagnose steht oft eine Odyssee von”. AerzteZeitung.de (на језику: немачки). 2004-12-15. Приступљено 2022-02-23.
- ^ Mayes, J. S.; Scheerer, J. B.; Sifers, R. N.; Donaldson, M. L. (1981). „Differential assay for lysosomal alpha-galactosidases in human tissues and its application to Fabry's disease”. Clinica Chimica Acta; International Journal of Clinical Chemistry. 112 (2): 247—251. PMID 6263521. doi:10.1016/0009-8981(81)90384-3.
- ^ Hoffmann, Bjoern; Georg Koch, Hans; Schweitzer-Krantz, Susanne; Wendel, Udo; Mayatepek, Ertan (2005). „Deficient α-galactosidase a activity in plasma but no Fabry disease – a pitfall in diagnosis”. Clinical Chemistry and Laboratory Medicine (CCLM). 43 (11): 1276—1277. PMID 16232095. S2CID 85843657. doi:10.1515/CCLM.2005.219..
- ^ Linthorst, Gabor E.; Vedder, Anouk C.; Aerts, Johannes M.F.G.; Hollak, Carla E.M. (2005). „Screening for Fabry disease using whole blood spots fails to identify one-third of female carriers”. Clinica Chimica Acta. 353 (1): 201—203. PMID 15698608. doi:10.1016/j.cccn.2004.10.019..
- ^ а б B. E. Smid, L. van der Tol, M. Biegstraaten, G. E. Linthorst, C. E. M. Hollak, B. J. H. M. Poorthuis: Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease. In: J Med Genet. Band 52, Nr. 4, 2015, S. 262–226.
- ^ Roy, S.; Gaudin, K.; Germain, D.P.; Baillet, A.; Prognon, P.; Chaminade, P. (2004). „Optimisation of the separation of four major neutral glycosphingolipids: Application to a rapid and simple detection of urinary globotriaosylceramide in Fabry disease”. Journal of Chromatography B. 805 (2): 331—337. PMID 15135109. doi:10.1016/j.jchromb.2004.03.037.
- ^ Desnick, Robert J. (2007). „Prenatal diagnosis of Fabry disease”. Prenatal Diagnosis. 27 (8): 693—694. PMID 17533632. S2CID 33020619. doi:10.1002/pd.1767..
- ^ A. M. Das: Angeborene Stoffwechselstörungen. In: A. Rieder, B. Lohff (eds.): Gender Medizin. Verlag Springer. Rieder, Anita; Lohff, Brigitte (2004). Gender Medizin: Geschlechtsspezifische Aspekte für die klinische Praxis. Springer. стр. 67f. ISBN 3-211-00766-0..
- ^ а б Kasper, David C.; Herkner, Kurt R.; Streubel, Berthold; Item, Chike B.; Ratschmann, Rene; Herman, Joseph L.; Shushan, Bori; Martin, Monica; Orsini, Joseph J.; Mechtler, Thomas P.; Metz, Thomas F. (2011). „Simplified Newborn Screening Protocol for Lysosomal Storage Disorders”. Clinical Chemistry. 57 (9): 1286—1294. PMID 21771947. doi:10.1373/clinchem.2011.164640..
- ^ M. Spada, D. Kasper, V. Pagliardini, E. Biamino, S. Giachero, F. Porta: Metabolic progression to clinical phenotype in classic Fabry disease. In: Ital J Pediatr. Band 43, Nr. 1, 2017, S. 1.
- ^ Pastores, G. M. (2006). „Miglustat: Substrate reduction therapy for lysosomal storage disorders associated with primary central nervous system involvement”. Recent Patents on CNS Drug Discovery. 1 (1): 77—82. PMID 18221193. doi:10.2174/157488906775245282.
- ^ „Therapie des Morbus Fabry”. 2015-02-03. Архивирано из оригинала 03. 02. 2015. г. Приступљено 2022-02-24.
- ^ а б Thomaidis, Thomas; Relle, Manfred; Reinke, Joerg; Beck, Michael; Schwarting, Andreas (2009). „Wirkung der Enzymersatztherapie (ERT) auf die Nierenfunktion von Patienten mit Morbus Fabry”. Medizinische Klinik. 104 (9): 699—703. PMID 19779674. S2CID 7132747. doi:10.1007/s00063-009-1152-1..
- ^ „March on, not in”. Nature Medicine. 17 (5): 515. 2011. PMID 21546944. S2CID 205378860. doi:10.1038/nm0511-515..
- ^ Beck, Michael (2002). „Agalsidase alfa – a preparation for enzyme replacement therapy in Anderson–Fabry disease”. Expert Opinion on Investigational Drugs. 11 (6): 851—858. PMID 12036428. S2CID 37196144. doi:10.1517/13543784.11.6.851.
- ^ Lee, K.; Jin, X.; Zhang, K.; Copertino, L.; Andrews, L.; Baker-Malcolm, J.; Geagan, L.; Qiu, H.; Seiger, K.; Barngrover, D.; McPherson, J. M.; Edmunds, T. (2003). „A biochemical and pharmacological comparison of enzyme replacement therapies for the glycolipid storage disorder Fabry disease”. Glycobiology. 13 (4): 305—313. PMID 12626384. doi:10.1093/glycob/cwg034..
- ^ Keating, G. M.; Simpson, D. (2007). „Spotlight on agalsidase beta in Fabry disease”. BioDrugs. 21 (4): 269—271. PMID 17628124. S2CID 26215578. doi:10.2165/00063030-200721040-00007.
- ^ Vedder, Anouk C.; Linthorst, Gabor E.; Houge, Gunnar; Groener, Johannna E.M.; Ormel, Els E.; Bouma, Berto J.; Aerts, Johannes M.F.G.; Hirth, Asle; Hollak, Carla E.M. (2007). „Treatment of Fabry Disease: Outcome of a Comparative Trial with Agalsidase Alfa or Beta at a Dose of 0.2 mg/Kg”. PLOS ONE. 2 (7): e598. Bibcode:2007PLoSO...2..598V. PMC 1913555 . PMID 17622343. doi:10.1371/journal.pone.0000598 .
- ^ Ohashi, Toya; Sakuma, Mio; Kitagawa, Teruo; Suzuki, Ken; Ishige, Nobuyuki; Eto, Yoshikatsu (2007). „Influence of antibody formation on reduction of globotriaosylceramide (GL-3) in urine from Fabry patients during agalsidase beta therapy”. Molecular Genetics and Metabolism. 92 (3): 271—273. PMID 17689998. doi:10.1016/j.ymgme.2007.06.013..
- ^ Arends, Maarten; Biegstraaten, Marieke; Wanner, Christoph; Sirrs, Sandra; Mehta, Atul; Elliott, Perry M.; Oder, Daniel; Watkinson, Oliver T.; Bichet, Daniel G.; Khan, Aneal; Iwanochko, Mark; Vaz, Frédéric M.; Van Kuilenburg, André B P.; West, Michael L.; Hughes, Derralynn A.; Hollak, Carla E M. (2018). „Agalsidase alfa versus agalsidase beta for the treatment of Fabry disease: An international cohort study”. Journal of Medical Genetics. 55 (5): 351—358. PMC 5931248 . PMID 29437868. S2CID 19054248. doi:10.1136/jmedgenet-2017-104863..
- ^ U. Fricke, U. Schwabe: Neue Arzneimittel 2007. In: U. Schwabe, D. Paffrath (eds.): Arzneiverordnungs-Report 2008. Verlag Springer, 2008, S. 74. eingeschränkte Vorschau in der Google-Buchsuche
- ^ Hoffmann, Björn; Mayatepek, Ertan (2009). „Fabry Disease”. Deutsches Ärzteblatt International. 106 (26): 440—447. PMC 270439 . PMID 19623315. doi:10.3238/arztebl.2009.0440.
- ^ Watt, Torquil; Burlina, Alessandro P.; Cazzorla, Chiara; Schönfeld, Dorothee; Banikazemi, Maryam; Hopkin, Robert J.; Martins, Ana Maria; Sims, Katherine; Beitner-Johnson, Dana; O'Brien, Fanny; Feldt-Rasmussen, Ulla (2010). „Agalsidase beta treatment is associated with improved quality of life in patients with Fabry disease: Findings from the Fabry Registry”. Genetics in Medicine. 12 (11): 703—712. PMID 20885332. S2CID 23666715. doi:10.1097/GIM.0b013e3181f13a4a..
- ^ Filling-Katz, M. R.; Merrick, H. F.; Fink, J. K.; Miles, R. B.; Sokol, J.; Barton, N. W. (1989). „Carbamazepine in Fabry's disease: Effective analgesia with dose-dependent exacerbation of autonomic dysfunction”. Neurology. 39 (4): 598—600. PMID 2494569. S2CID 38971405. doi:10.1212/wnl.39.4.598.
- ^ National Institute for Health and Clinical Excellence: Neuropathic pain: The pharmacological management of neuropathic pain in adults in non-specialist settings (NICE clinical guideline 96). Centre for Clinical Practice at NICE; 2010.
- ^ Cruccu, G.; Sommer, C.; Anand, P.; Attal, N.; Baron, R.; Garcia-Larrea, L.; Haanpaa, M.; Jensen, T. S.; Serra, J.; Treede, R. -D. (2010). „EFNS guidelines on neuropathic pain assessment: Revised 2009”. European Journal of Neurology. 17 (8): 1010—1018. PMID 20298428. S2CID 2393393. doi:10.1111/j.1468-1331.2010.02969.x..
- ^ Argoff, C. E.; Barton, N. W.; Brady, R. O.; Ziessman, H. A. (1998). „Gastrointestinal symptoms and delayed gastric emptying in Fabry's disease: Response to metoclopramide”. Nuclear Medicine Communications. 19 (9): 887—891. PMID 10581595. doi:10.1097/00006231-199809000-00009.
- ^ Tahir, Hindia; Jackson, Leslie L.; Warnock, David G. (2007). „Antiproteinuric Therapy and Fabry Nephropathy”. Journal of the American Society of Nephrology. 18 (9): 2609—2617. PMID 17656478. doi:10.1681/ASN.2006121400..
- ^ Stetter, Christian E. (2007). In vivo Untersuchung des kardialen Stoffwechsels bei Morbus Fabry mittels 31Phosphor-MR-Spektroskopie (Теза). Universität Würzburg.
Литература
уреди- Tucić, N, Matić, Gordana: O genima i ljudima, Centar za primenjenu psihologiju, Beograd, 2002.
- Marinković, D, Tucić, N, Kekić, V: Genetika, Naučna knjiga, Beograd
- Tatić, S, Kostić, G, Tatić, B: Humani genom, ZUNS, Beograd, 2002.
- Matić, Gordana: Osnovi molekularne biologije, Zavet, Beograd, 1997.
- Ridli, M: Genom - autobiografija vrste u 23 poglavlja, Plato, Beograd, 2001.
- Prentis S: Biotehnologija, Školska knjiga, Zagreb, 1991.
- Dumanović, J, marinković, D, Denić, M: Genetički rečnik, Beograd, 1985.
- Kosanović, M, Diklić, V: Odabrana poglavlja iz humane genetike, Beograd, 1986.
- Švob, T. i sradnici: Osnovi opće i humane genetike, Školska knjiga, Zagreb, 1990.
Спољашње везе
уреди| Класификација | |
|---|---|
| Спољашњи ресурси |
- „Fabry Disease Information Page”. Архивирано из оригинала 02. 12. 2016. г. at NINDS (језик: енглески)
- Fabry disease at NLM Genetics Home Reference (језик: енглески)
 | Молимо Вас, обратите пажњу на важно упозорење у вези са темама из области медицине (здравља). |