Aromatično jedinjenje
U organskoj hemiji, strukture aromatičnih jedinjenja su neuobičajeno stabilne. Aromatičnost je hemijska osobina kojom konjugovani prsten nezasićenih veza, slobodnih parova, ili praznih orbitala pokazuje stabilizaciju jaču nego što bi se očekivalo kao posledica same konjugacije. To se takođe smatra manifestacijom ciklične delokalizacije i rezonance.[1][2][3]

Obično se smatra da je to zato što su elektroni slobodni da se kreću oko kružnog aranžmana atoma koji su alternativno jednostruko- i dvostruko-vezani jedan za drugog. Na te veze se može gledati kao hibrid jednostrukih i dvostrukih veza, gde je sve veza u prstenu identične. Ovaj uobičajeni model aromatičnog prstena, zapravo ideju da je benzen formiran od šestočlanog ugleničnog prstena sa naizmeničnim jednostrukim i dvostrukim vezama (cikloheksatrien), je razvio Fridrih Kekule. Model benzena se sastoji od dve rezonantne forme. Benzen je stabilniji molekul nego što bi se očekivalo bez uzimanja u obzir delokalizacije naelektrisanja.
Teorija
уреди
Kao što je standardno za rezonantne dijagrame, dvoglava strela se koristi da se ukaže da dve strukture nisu distinktni entiteti, nego samo hipotetične mogućnosti. Nijedna od njih nije precizna reprezentacija stvarnog jedinjenja, koje je najbolje prikazano kao hibrid (prosek) tih struktura, što se može videti sa leve strane. C=C je kraća od C−C veze, dok je benzen perfektno heksagonalan — svih šest ugljenik-ugljenik veza imaju istu dužinu, koja je između jednostruke i dvostruke veze.[4][5]
Bolja reprezentacija je ta sa cirkularnom π vezom (Armstrongov unutrašnji krug), u kojoj je elektronska gustina ravnomerno distribuirana kroz π-veze iznad i ispod prstena. Ova model korektnije predstavlja lokaciju elektronske gustine unutar aromatičnog prstena.
Jednostruke veze su formirane sa elektronima u liniji između dva ugljenika. One se nazivaju σ-vezama. Dvostruke veze se sastoje of σ-veze i π-veze. π-veze se formiraju preklapanjem atomskih p-orbitala iznad i ispod ravni prstena. Sledeći dijagram pokazuje pozicije tih p-orbitala:

Pošto su one van ravni atoma, te orbitale mogu slobodno da interaguju jedna s drugom, i da postanu delokalizovane. To znači da umesto da su vezeni za jedan atom ugljenika, svi elektroni se deli između svih šest atoma u prstenu. Pošto, nema dovoljno elektrona da se formiraju dvostruke veze na svim ugljenikovim atomima, nego "ekstra" elektroni pojačavaju sve veze prstena podjednako. Rezultujuća molekularska orbitala ima π simetriju.

Istorija
уредиTermin „aromatičan”
уредиPrva pozanta upotreba reči „aromatičan” kao hemijskog termina, za označavanje jedinjenja koja sadrže fenil grupu, javila se u članku koji je objavio August Vilhelm Hofman 1855. godine.[6][7] Ako je ovo zaista najranije uvođenje termina, zanimljivo je da Hofman ne kaže nište o razlogu iz kog je uveo ovaj pridev koji ukazuje na mirisno svojstvo za opisivanje grupe hemijskih supstanci od kojih samo neke imaju primetne arome. Isto tako, mnoge od organskih supstanci jakog mirisa poznatih kao terpeni, nisu aromatična u hemijskom smislu. Terpeni i benzenoidne supstance doduše imaju zajedničke hemijske karakteristike, kao što je viši stepen nezasićenosti od većine alifatičnih jedinjenja, i moguće je da je Hofman želeo da napravi razliku između te dve kategorije. Mnoga rano poznata aromatična jedinjenja, kao što su benzen i toluen, imala su prepoznatljive prijatne mirise. Ovo svojstvo je verovatno dovelo do toga da se termin „aromatičan” koristi za ovu klasu jedinjenja, i stoga termin „aromatičnost” za elektronsko svojstvo koje je kasnije otkriveno.[8]
Struktura benzenskog prstena
уреди.png)

U 19. veku hemičari su smatrali da je zbunjujuće da benzen može biti tako neaktivan u pogledu reakcije adicije, imajući u vidu njegov pretpostavljeni visoki stupanj nezasićenosti. Cikloheksatriensku strukturu benzena je prvi predložio Fridrih Kekule 1865. godine.[10][11] Većina hemičara je brzo prihvatila ovu strukturu, jer je ona zadovoljavala većinu poznatih izomernih odnosa aromatične hemije. Heksagonalna struktura objašnjava zašto postoji samo jedan izomer benzena i zašto disupstituisana jedinjenja imaju tri izomera.[7]
Između 1897. i 1906, Dž. Dž. Tomson, pronalazač elektrona je predložio tri ekvivalentna elektrona između svakog para atoma ugljenika u benzenu. Zasluge za objašnjavanje izuzetne stabilnosti benzena se konvencionalno pripisuju Ser Robertu Robinsonu, koji je prvi (1925. godine)[12] skovao termin aromatični sekstet za grupu od šest elektrona otpornih na poremećaje.
Zapravo, ovaj koncept se može pratiti dalje u prošlost, preko Ernesta Krokera iz 1922,[13] do Henrija Edvarda Armstronga, koji je 1890. godine pisao „(šest) centričnih afiniteta deluju unutar kruga ... benzen se može predstaviti sa dvostrukim prstenom (sic) ... i kad se aditivno jedinjenje formira, unutarnji krug afiniteta trpi poremećaj, susedni atomi ugljika na koje ništa nije vezano nužno stiču etilensko stanje”.[14] Ovde je Armstrong opisivao bar četiri moderna koncepta. Prvo, njegov „afinitet” je u današnje vreme poznatiji kao elektron, kojeg je otkrio samo sedam godina kasnije Dž. Dž. Tomson. Drugo, on opisuje elektrofilnu aromatičnu supstituciju, prolazeći (treće) kroz Velandov intermedijar, u kome je (četvrto) konjugacija prstena narušena. On je uveo simbol C centriran na prstenu kao oznaku za unutrašnji krug, tako anticipirajući notaciju Eriha Klara. Tvrdi se da je on takođe predvideo prirodu talasne mehanike, budući da je prepoznao da njegovi afiniteti imaju smer, da nisu samo tačkaste čestice, i da kolektivno imaju distribuciju koja se može promeniti uvođenjem supstituenata na benzenski prsten (kao što se distribucija električnog naboja u telu menja kad mu se približava drugo telo).
Kvantno mehaničko poreklo ove stabilnosti, ili aromatičnosti, prvi je modelovao Hikel 1931. godine. On je prvi razdvojio vezujuće elektrone u sigma i pi elektrone.
Aromatičnost jednog proizvoljnog aromatičnog jedinjenja se može kvantitativno meriti pomoću računarskog metoda hemijskog pomeranja nezavisnog od jezgra (engl. Nucleus-Independent Chemical Shift - NICS)[15] i metoda procenta aromatičnosti.[16]
Tipovi aromatičnih jedinjenja
уредиVelika većina aromatičnih jedinjenja su jedinjenja ugljenika, mada ta jedinjenja ne moraju da budi ugljovodonici.
Heterociklična jedinjenja
уредиKod heterocikličnih aromatičnih jedinjenja (heteroaromata), jedan ili više atoma u aromatičnom prstenu nije ugljenik. To može da umanji aromatičnost prstena, i tako (kao što je to slučaj sa furanom) poveća njegovu reaktivnost. Drugi primeri su piridin, pirazin, imidazol, pirazol, oksazol, tiofen, i njihovi benzanilisani analozi (npr. benzimidazol). Aromataska hreterociklična jedinjenja su heterociklusi koji ispunjavaju kriterij aromatičnosti:imaju planarnu strukturu, neprekinuti niz p elektrona, te neparan broj parova p elektrona u ciklusu(dvostruke veze i usamljeni elektronski parovi. Najznačajniji su oni sa 5 ili 6 atoma u ciklusu.
Policiklična jedinjenja
уредиPoliciklični aromatični ugljovodonici su molekuli koji sadrže dva ili više jednostavna aromatična prstena vezan putem zajedničke veze, i.e. prsteni dele dva atoma. Primeri su naftalen, antracen i fenantren.
Supstituisani aromatici
уредиPostoji veliki broj hemijskih jedinjenja koja su aromatični prstenovi sa jednim ili više vezanih supstituenata. Primeri su trinitrotoluen (TNT), acetilsalicilna kiselina (aspirin), paracetamol, i DNK nukleotidi.
Atipična aromatična jedinjenja
уредиAromatičnost je isto tako nađena kod jona: ciklopropenilni katjon (2e sistem), ciklopentadienilni anjon (6e sistem), tropilijumski jon (6e) i ciklooktatetraenski dianjon (10e). Aromatične osobine su bile pripisane ne-benzenoidnim jedinjenjima kao što je tropon. Granični slučaj aromatičnih osobina poseduje klasa jedinjenja zvanih ciklofani.
Heteroareni
уредиHeteroareni su aromatična jedinjenja, gde je najmanje jedna metilenska ili vinilenska (-C= ili -CH=CH-) grupa zamenjena heteroatomom: kiseonikom, azotom ili sumporom.[17] Primeri nebenzenskih jedinjenja sa aromatičnim svojstvima su furan, heterociklično jedinjenje sa petočlanim prstenom koji uključuje jedan atom kiseonika, i piridin, heterociklično jedinjenje sa šestočlanim prstenom koje sadrži jedan atom azota. Ugljovodonici bez aromatičnog prstena nazivaju se alifatičnim. Otprilike polovina jedinjenja poznatih 2000. godine opisana je kao aromatična u izvesnoj meri.[18]



Aplikacije
уредиAromatična jedinjenja su rasprostranjena u prirodi i industriji. Ključni industrijski aromatični ugljovodonici su benzol, toluen, ksilen koji se naziva BTX. Mnogi biomolekuli imaju fenil grupe uključujući takozvane aromatične aminokiseline.
Model benzenskog prstena
уреди

Benzen, C6H6, je najmanje složen aromatični ugljovodonik, i bio je prvi koji je definisan kao takav.[20] Njegovu prirodu vezivanja prvi su nezavisno prepoznali Džozef Lošmit i Avgust Kekule u 19. veku.[20] Svaki atom ugljenika u heksagonalnom prstenu ima četiri elektrona da podeli. Jedan elektron formira sigma vezu sa atomom vodonika, a jedan se koristi u kovalentnom vezivanju za svaki od dva susedna ugljenika. Ovo ostavlja šest elektrona, podjednako podeljenih oko prstena u delokalizovanim pi molekularnim orbitalama veličine samog prstena.[19] Ovo predstavlja šest ekvivalentnih veza ugljenik-ugljenik sve od kojih su 1,5 reda veze. Ova ekvivalencija se takođe može objasniti rezonantnim oblicima.[19] Elektroni su vizualizovani kao lebdeći iznad i ispod prstena, a elektromagnetna polja koja generišu deluju tako da drže prsten u ravni.[19]
Simbol kruga za aromatičnost uveli su ser Robert Robinson i njegov učenik Džejms Armit 1925. godine, a popularizovali su ga počev od 1959. Morison & Bojd svojim udžbenikom organske hemije.[21] O pravilnoj upotrebi simbola se raspravlja: neke publikacije ga koriste za bilo koji ciklični π sistem, dok ga druge koriste samo za one π sisteme koji se povinjavaju Hikelovom pravilu. Neki tvrde da bi, kako bi se ostalo u skladu sa Robinsonovim prvobitno iznetim predlogom, upotreba simbola kruga trebalo da bude ograničena na monociklične 6 π-elektronske sisteme.[22] Na ovaj način se simbol kruga za vezu sa šest centara sa šest elektrona može uporediti sa Y simbolom za dvoelektronsku tricentarsku vezu.[22]
Benzen i derivati benzola
уреди
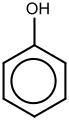
Derivati benzena imaju od jedan do šest supstituenata vezanih za centralno jezgro benzena. Primeri jedinjenja benzena sa samo jednim supstituentom su fenol, koji nosi hidroksilnu grupu, i toluen sa metil grupom. Kada je na prstenu prisutno više od jednog supstituenta, njihov prostorni odnos postaje važan za šta su osmišljeni obrasci supstitucije arena orto, meta i para.[23] Kada reaguju da bi se formirali složeniji derivati benzena, supstituenti na benzenskom prstenu se mogu opisati kao aktivirani ili deaktivirani, a to su davanje elektrona, odnosno povlačenje elektrona.[23] Aktivatori su poznati kao orto-para direktori, a deaktivatori su poznati kao meta direktori.[23] Nakon reakcije, supstituenti će biti dodati na orto, para ili meta pozicijama, u zavisnosti od usmerenosti trenutnih supstituenata da bi se dobili složeniji derivati benzena, često sa nekoliko izomera. Protok elektrona koji vodi do rearomatizacije je ključan u obezbeđivanju stabilnosti takvih proizvoda.[23]
Na primer, postoje tri izomera za krezol jer se metil grupa i hidroksilna grupa (oba orto para usmerivači) mogu postaviti jedan pored drugog (orto), jedna pozicija razmaka jedno od drugog (meta) ili dve pozicije razmaka jedno od drugog (para).[24] S obzirom da su metil i hidroksilna grupa orto-para usmerivači, orto i para izomeri su tipično favorizovani.[24] Ksilenol ima dve metil grupe pored hidroksilne grupe, i za ovu strukturu postoji 6 izomera.
- Reprezentativna jedinjenja arena
-

-

-

-

-

-

-

-

-
-

-

-

-

-

-














Arenski prstenovi mogu stabilizovati naelektrisanja, kao što se vidi, na primer, u fenolu (C6H5–OH), koji je kiseo na hidroksilu (OH), pošto je naelektrisanje kiseonika (alkoksid –O−) delimično delokalizovano u benzenski prsten.
Nebenzilni areni
уредиIako su benzil areni uobičajeni, nebenzilna jedinjenja su takođe izuzetno važna. Svako jedinjenje koje sadrži ciklični deo koji je u skladu sa Hikelovim pravilom i nije derivat benzena može se smatrati nebenzilnim aromatičnim jedinjenjem.[19]
Monociklični areni
уредиOd anulena većih od benzena, [12]anulen i [14]anulen su slabo aromatična jedinjenja, a [18]anulen, ciklooktadekanonaen je aromatičan, iako naprezanje unutar strukture izaziva blago odstupanje od tačno ravne strukture neophodne za aromatičnu kategorizaciju.[25] Drugi primer nebenzilnog monocikličnog arena je ciklopropenil (ciklopropenijum katjon), koji zadovoljava Hikelovo pravilo sa n jednakim 0.[26] Treba imati na umu da je samo katjonski oblik ovog cikličnog propenila aromatičan, s obzirom da bi neutralnost u ovom jedinjenju prekršila bilo oktetno pravilok ili Hikelovo pravilo.[26]
Drugi nebenzilni monociklični areni uključuju gorepomenute heteroarene koji mogu zameniti atome ugljenika drugim heteroatomima kao što su N, O ili S.[19] Uobičajeni primeri su šestočlani pirol i petočlani piridin, od kojih oba imaju supstituisani azot.[27]
Policiklični aromatični ugljovodonici
уреди
Policiklični aromatični ugljovodonici, takođe poznati kao polinuklearna aromatična jedinjenja (PAH) su aromatični ugljovodonici koji se sastoje od kondenzovanih aromatičnih prstenova i ne sadrže heteroatome, niti nose supstituente.[28] Naftalen je najjednostavniji primer PAH-a. PAH jedinjenja se javljaju u nalazištima nafte, uglja i katrana i proizvode se kao nusproizvodi sagorevanja goriva (bilo da se radi o fosilnom gorivu ili biomasi).[29] Kao zagađivači, oni su uzrok zabrinutosti jer su neka jedinjenja identifikovana kao kancerogena, mutagena i teratogena.[30][31][32][33] PAH jedinjenja se takođe nalaze u termičkih obrađenoj hrani.[29] Studije su pokazale da se visoki nivoi PAH-a nalaze, na primer, u mesu kuvanom na visokim temperaturama kao što je pečenje na roštilju, i u dimljenoj ribi.[29][30] Oni su takođe dobri kandidati za molekule da deluju kao osnova za najranije oblike života.[34] U grafenu je PAH motiv proširen na velike 2D listove.[35]
Vidi još
уредиReference
уреди- ^ Schleyer, Paul von Ragué (2001). „Introduction: Aromaticity”. Chemical Reviews. 101 (5): 1115. PMID 11749368. doi:10.1021/cr0103221.
- ^ A. T. Balaban, P. v. R. Schleyer and H. S. Rzepa (2005). „Crocker, Not Armit and Robinson, Begat the Six Aromatic Electrons”. Chemical Reviews. 105: 3436—3447. doi:10.1021/cr0300946.
- ^ Schleyer, Paul von Ragué (2005). „Introduction: DelocalizationPi and Sigma”. Chemical Reviews. 105: 3433. doi:10.1021/cr030095y.
- ^ „Bonding in benzene – the Kekulé structure”. www.chemguide.co.uk. Приступљено 25. 12. 2015.
- ^ „Chemical Reactivity”. www2.chemistry.msu.edu. Приступљено 25. 12. 2015.
- ^ Hofmann, A. W. (1855). „On Insolinic Acid”. Proceedings of the Royal Society. 8: 1—3. doi:10.1098/rspl.1856.0002.
- ^ а б Rocke, A. J. (2015). „It Began with a Daydream: The 150th Anniversary of the Kekulé Benzene Structure”. Angew. Chem. Int. Ed. 54: 46—50. PMID 25257125. doi:10.1002/anie.201408034.
- ^ McMurry, John (2007). Organic Chemistry (7th изд.). Brooks-Cole. стр. 515. ISBN 978-0-495-11258-7.
- ^ Kekulé, F. A. (1872). „Ueber einige Condensationsproducte des Aldehyds”. Liebigs Ann. Chem. 162 (1): 77—124. doi:10.1002/jlac.18721620110.
- ^ Kekulé, F. A. (1865). „Sur la constitution des substances aromatiques”. Bulletin de la Societe Chimique de Paris. 3: 98—110.
- ^ Kekulé, F. A. (1866). „Untersuchungen über aromatische Verbindungen Ueber die Constitution der aromatischen Verbindungen. I. Ueber die Constitution der aromatischen Verbindungen”. Liebigs Ann. Chem. 137 (2): 129—196. doi:10.1002/jlac.18661370202.
- ^ Armit, James Wilson; Robinson, Robert (1925). „CCXI. Polynuclear heterocyclic aromatic types. Part II. Some anhydronium bases”. J. Chem. Soc. Trans. 127: 1604—1618. doi:10.1039/CT9252701604.
- ^ Crocker, Ernest C. (1922). „Application Of The Octet Theory To Single-Ring Aromatic Compounds”. J. Am. Chem. Soc. 44 (8): 1618—1630. doi:10.1021/ja01429a002.
- ^ Armstrong, Henry Edward (1890). „The structure of cycloid hydrocarbon”. Proc. Chem. Soc. 6 (85): 95—106. doi:10.1039/PL8900600095.
- ^ Schleyer, Paul von Ragué; Maerker, Christoph; Dransfeld, Alk; Jiao, Haijun; Van Eikema Hommes, Nicolaas J. R. (1996). „Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe”. J. Am. Chem. Soc. 118 (26): 6317—6318. doi:10.1021/ja960582d.
- ^ Mucsi, Z.; Viskolcz, B.; Csizmadia, I. G. (2007). „A Quantitative Scale for the Degree of Aromaticity and Antiaromaticity”. J. Phys. Chem. A. 111 (6): 1123—1132. Bibcode:2007JPCA..111.1123M. PMID 17286363. doi:10.1021/jp0657686.
- ^ IUPAC. Compendium of Chemical Terminology, 2nd ed. (the "Gold Book"). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). Online version (2019-) created by S. J. Chalk. ISBN 0-9678550-9-8. https://doi.org/10.1351/goldbook.
- ^ Balaban, Alexandru T.; Oniciu, Daniela C.; Katritzky, Alan R. (2004-05-01). „Aromaticity as a Cornerstone of Heterocyclic Chemistry”. Chemical Reviews (на језику: енглески). 104 (5): 2777—2812. ISSN 0009-2665. PMID 15137807. doi:10.1021/cr0306790.
- ^ а б в г д ђ е ж з и ј Klein, David R. (2017). Organic Chemistry (3rd изд.). John Wiley & Sons. ISBN 9781119444251.
- ^ а б „Benzene | Definition, Discovery, Structure, Properties, & Uses | Britannica”. www.britannica.com (на језику: енглески). Приступљено 2023-11-06.
- ^ Armit, James Wilson; Robinson, Robert (1925). „CCXI.—Polynuclear heterocyclic aromatic types. Part II. Some anhydronium bases”. J. Chem. Soc., Trans. (на језику: енглески). 127: 1604—1618. ISSN 0368-1645. doi:10.1039/CT9252701604.
- ^ а б Jensen, William B. (април 2009). „The Origin of the Circle Symbol for Aromaticity”. Journal of Chemical Education (на језику: енглески). 86 (4): 423. Bibcode:2009JChEd..86..423J. ISSN 0021-9584. doi:10.1021/ed086p423.
- ^ а б в г „16.5: An Explanation of Substituent Effects”. Chemistry LibreTexts (на језику: енглески). 2015-05-03. Приступљено 2023-12-03.
- ^ а б „Cresol - an overview | ScienceDirect Topics”. www.sciencedirect.com. Приступљено 2023-12-03.
- ^ „What does "aromatic" really mean?”. Chemistry LibreTexts (на језику: енглески). 2013-10-02. Приступљено 2023-11-06.
- ^ а б „What does "aromatic" really mean?”. Chemistry LibreTexts (на језику: енглески). 2013-10-02. Приступљено 2023-11-29.
- ^ „4.2: Covalent Bonds”. Chemistry LibreTexts (на језику: енглески). 2020-07-30. Приступљено 2023-11-06.
- ^ Fetzer, John C. (2007-04-16). „THE CHEMISTRY AND ANALYSIS OF LARGE PAHs”. Polycyclic Aromatic Compounds (на језику: енглески). 27 (2): 143—162. ISSN 1040-6638. S2CID 97930473. doi:10.1080/10406630701268255.
- ^ а б в "Polycyclic Aromatic Hydrocarbons – Occurrence in foods, dietary exposure and health effects" (PDF). European Commission, Scientific Committee on Food. December 4, 2002. Archived (PDF) from the original on 2022-10-09.
- ^ а б Larsson, Bonny K.; Sahlberg, Greger P.; Eriksson, Anders T.; Busk, Leif A. (јул 1983). „Polycyclic aromatic hydrocarbons in grilled food”. Journal of Agricultural and Food Chemistry (на језику: енглески). 31 (4): 867—873. ISSN 0021-8561. PMID 6352775. doi:10.1021/jf00118a049.
- ^ Scientific Opinion of the Panel on Contaminants in the Food Chain on a request from the European Commission on Marine Biotoxins in Shellfish – Saxitoxin Group. The EFSA Journal (2009) 1019, 1-76.
- ^ Keith, Lawrence H. (2015-03-15). „The Source of U.S. EPA's Sixteen PAH Priority Pollutants”. Polycyclic Aromatic Compounds (на језику: енглески). 35 (2–4): 147—160. ISSN 1040-6638. doi:10.1080/10406638.2014.892886.
- ^ Thomas, Philippe J.; Newell, Emily E.; Eccles, Kristin; Holloway, Alison C.; Idowu, Ifeoluwa; Xia, Zhe; Hassan, Elizabeth; Tomy, Gregg; Quenneville, Cheryl (2021-02-01). „Co-exposures to trace elements and polycyclic aromatic compounds (PACs) impacts North American river otter (Lontra canadensis) baculum”. Chemosphere. 265: 128920. ISSN 0045-6535. doi:10.1016/j.chemosphere.2020.128920
 .
.
- ^ Ehrenfreund, Pascale; Rasmussen, Steen; Cleaves, James; Chen, Liaohai (јун 2006). „Experimentally Tracing the Key Steps in the Origin of Life: The Aromatic World”. Astrobiology (на језику: енглески). 6 (3): 490—520. Bibcode:2006AsBio...6..490E. ISSN 1531-1074. PMID 16805704. doi:10.1089/ast.2006.6.490.
- ^ Wang, Xiao-Ye; Yao, Xuelin; Müllen, Klaus (2019-09-01). „Polycyclic aromatic hydrocarbons in the graphene era”. Science China Chemistry (на језику: енглески). 62 (9): 1099—1144. ISSN 1869-1870. S2CID 198333072. doi:10.1007/s11426-019-9491-2 . hdl:21.11116/0000-0004-B547-0 .
Literatura
уреди- R.L. Shriner; C.K.F. Hermann; T.C. Morrill; D.Y. Curtin & R.C. Fuson John (1997). The Systematic Identification of Organic Compounds. Wiley & Sons. ISBN 978-0-471-59748-3.
- Richard F. & Daley, Sally J. Organic Chemistry, Online organic chemistry textbook.
- Roberts, John D.; Caserio, Marjorie C. (1964). Basic Principles of Organic Chemistry. W. A. Benjamin, Inc.
- Streitwieser, Andrew; Heathcock, Clayton H.; Kosower, Edward M. (2017). Introduction to Organic Chemistry. New Delhipages=3–4: Medtech (Scientific International, reprint of revised 4th edition, Macmillan, 1998). ISBN 978-93-85998-89-8.
- Henry Marshall Leicester; Herbert S. Klickstein (1951). A Source Book in Chemistry, 1400-1900. Harvard University Press. стр. 309.
- Nicolaou, K.C.; Sorensen, E.J. (1996). Classics in Total Synthesis: Targets, Strategies, Methods. Wiley. ISBN 978-3-527-29231-8.
Spoljašnje veze
уреди- MIT.edu, OpenCourseWare: Organic Chemistry I
- HaverFord.edu, Organic Chemistry Lectures, Videos and Text
- Organic-Chemistry.org, Organic Chemistry Portal – Recent Abstracts and (Name)Reactions
- Orgsyn.org, Organic Chemistry synthesis journal
- Khanacademy.org, Khan Academy - Organic Chemistry